
Наследственный нефрит или синдром Альпорта – это генетическое заболевание, которое встречается нечасто. Признаки патологии проявляются у детей от 3-х лет до достижения ими младшего школьного возраста. Болезнь чаще встречается у мальчиков. В некоторых случаях она длительное время может не выражаться яркими симптомами, поэтому выявляется при появлении подозрений на другие заболевания во время проведения диагностических исследований или медицинских осмотров.
Содержание
Причины возникновения
Генетическое изменение гена, отвечающего за процесс биосинтеза коллагена – это основная предпосылка к развитию синдрома Альпорта. Такую наследственную мутацию ребёнок приобретает от родителей: дочь – от отца, а от матери – как мальчик, так и девочка. У человека может быть поврежден ген, при этом болезнь развивается не всегда, но будет наследоваться детьми.
Изменению подвергается один из трех генов, отвечающих за процесс биологического синтеза коллагена типа IV. Именно он входит в структуру базальных мембран, расположенных в почках, органах зрения и слуха. Базальные мембраны являются особыми образованиями – тоненькими границами, которыми некоторые ткани организма человека отделяются друг от друга, поддерживают и укрепляют их структуру. Если их состав и целостность нарушаются, возникают опасные явления:
- происходит неполная и некачественная фильтрация токсических и других веществ, поступающих из крови;
- изменяется состав мочи, в которой наблюдается значительное увеличение эритроцитов (гематурия или следы крови в моче) и нефильтрованного белка (протеинурия).
Такие изменения приводят к развитию почечной недостаточности тяжелого типа, в некоторых случаях к полному прекращению функций органа и летальному исходу.

Синдром Альпорта и его развитие могут спровоцировать некоторые факторы:
- тяжелые заболевания, вызванные бактериями или вирусами;
- воздействие на организм компонентов вакцин;
- чрезмерные нагрузки физического характера.
Медицинская статистика обладает данными о том, что каждый пятый ребенок с подтвержденным диагнозом наследственный нефрит, появляется у родителей, не имеющих патологий, связанных с работой почек. Причина возникновения синдрома Альпорта в таких случаях связана с генными мутациями спонтанного типа.
Классификация и характерные признаки
Генетический нефрит имеет две классификации. Специалисты выделяют его виды в зависимости от типа наследования патологии и особенностей развития заболевания.
Различают три разновидности наследственного нефрита:
- классический или Х-сцепленный доминантный – является наиболее распространенным, подтверждается у 80 % пациентов;
- аутосомно-рецессивный – ему подвержены не более 15 % больных;
- аутосомно доминантный синдром Альпорта – встречается в медицинской практике крайне редко, диагностируется менее чем у 5 % пациентов.
Врач-нефролог классифицирует диагноз синдром Альпорта в зависимости от особенностей проявлений патологии:
- изменения работы почек, предполагающие наличие гематурии с одновременными нарушениями зрения и слуха;
- нефрит с признаками гематурии, развивающийся без снижения функций органов зрения и слуха;
- семейная гематурия доброкачественного характера, не имеющая признаков хронической почечной недостаточности.
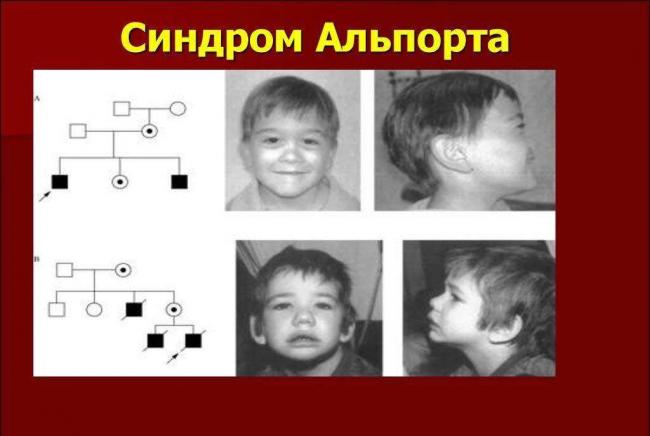
Так как причиной возникновения синдрома Альпорта является нарушение выработки основного элемента соединительных тканей – коллагена, главные его симптомы связаны с процессами изменения строения базальных мембран. Их структура нарушается в важных отделах организма человека:
- почечных клубочках;
- внутреннем ухе;
- органах зрения.
Самым распространённым и первично возникающим симптомом является наличие гематурии. Кровь в моче может быть видимой или выявляется при лабораторных исследованиях. Появление признаков тугоухости, полной односторонней глухоты (в некоторых случаях), ухудшение зрения усиливают подозрения на возможное развитие синдрома Альпорта. У пациентов в моче может подтверждаться наличие белка.
Когда у ребенка развивается нефрит наследственного характера, его состояние свидетельствует об интоксикации организма, которую можно определить по следующим симптомам:
- снижение уровня гемоглобина;
- головокружение, шум в ушах;
- частое появление и повторение мышечной и головной боли;
- изменение дыхания, которое становится поверхностным;
- наличие одышки;
- значение артериального давления не является стабильным, оно может как повышаться, так и понижаться;
- появление бессонницы или чрезмерной сонливости.
В случаях развития наследственного нефрита в хронической форме состояние пациента усугубляется проявлением дополнительных признаков:
- проблемы с процессом мочеиспускания, увеличение частоты посещений туалета;
- наличие примеси крови в моче;
- признаки общего недомогания, сильная слабость, снижение аппетита;
- наличие мышечных судорог;
- появление кровоподтеков, кожного зуда;
- заметные болевые ощущения в области груди;
- изменение сознания, иногда до беспамятства.
Патология может протекать с внешними признаками. У ребенка возможно особое строение ушных раковин и нёба, срастание пальцев или наличие дополнительных, выраженная складка у внутреннего угла глаза.
Диагностика
Поставить диагноз наследственного нефрита помогает проведение нескольких исследований, сбор и анализ анамнеза. Именно генетический характер болезни предполагает изучение особенностей здоровья родителей, бабушек и дедушек малыша. Необходима оценка показателей, наличие трех из них позволяет диагностировать синдром:
- признаки гематурии;
- патологии почек, почечная недостаточность, в том числе закончившаяся смертью;
- заболевания органов зрения и слуха врожденного характера;
- прогрессирующее ухудшение качества зрительного и слухового восприятия у ребенка;
- наличие изменений строения базальной мембраны (проводится при биопсии почечной ткани).

Для оценки состояния пациента и особенностей развития патологического процесса врач назначает исследования:
- анализ на синдром Альпорта (предполагает изучение крови и мочи);
- ультразвуковое исследование почек, надпочечников;
- проведение рентгенографии органа;
- биопсия тканей;
- ДНК-тест для определения носителя гена-мутанта.
Пациенту требуются дополнительные посещения врачей узкой специализации – нефролога, генетика, окулиста, отоларинголога.
Необходимое лечение
Лечение синдрома Альпорта заключается в нормализации работы почек, так как особых, специфических медикаментов для избавления от всех симптомов патологии не существует.
Врач назначает лечение, уменьшающее риск прогрессирования синдрома:
- препараты, корректирующие проявления почечной недостаточности, уровень белка в моче;
- мочегонные средства;
- средства, нормализующие обмен веществ в организме, восстанавливающие баланс минералов и витаминов;
- препараты для профилактики анемии.
При выраженной почечной недостаточности показано использованием процедуры гемодиализа. В тяжелых случаях развития наследственного нефрита требуется замена пораженного органа на донорский. Трансплантация почки может быть выполнена пациентам в возрасте старше 15 лет.
Риск осложнений
Развитие почечной недостаточности является опасным осложнением синдрома Альпорта. Если вовремя не диагностировать болезнь и не приступить к её лечению, человек не проживет долго. Смерть с высокой долей вероятности настигнет его до достижения тридцатилетия.
Диетическое питание
Большое значение при диагнозе наследственный нефрит уделяется переходу на особый рацион питания. Диета при синдроме Альпорта предполагает полное исключение «неправильных» продуктов:
- с содержанием консервантов и ненатуральных пищевых добавок;
- острых и соленых блюд;
- жирных ингредиентов;
- с большим содержанием белка;
- любых алкогольных напитков.
В диете используются продукты, индивидуально рекомендованные врачом. Их список формируется с учетом функциональных способностей почек каждого пациента. Предлагается ввод в рацион компонентов, обеспечивающих больного необходимой энергией, витаминами и микроэлементами, а также обладающих достаточной калорийностью – телятина, нежирная говядина, птица, рыба, фрукты и овощи.
Прогноз и профилактика
В зависимости от классификации наследственного нефрита прогноз может значительно отличаться. Наибольшему риску возможного летального исхода подвержены пациенты, у которых прогрессирует хроническая почечная недостаточность, наблюдаемая при высокой степени протеинурии. В этом списке чаще находятся мальчики. В большинстве случаев пациентам необходима пересадка почки.
Если синдром имеет аутосомно-доминантную форму, прогноз при выполнении всех врачебных рекомендаций благоприятный. В случаях выявления семейной гематурии доброкачественного характера требуется лечение, предупреждающее развитие симптомов.
Воспитание и обучение детей с синдромов Альпорта предполагает особый подход, многим из них рекомендуют получение образования в домашних условиях. Такой вид обучения не допускает значительных физических и нервных нагрузок, позволяет учитывать возможные изменения слухового и зрительного восприятия ребенка. Если состояние малыша позволяет посещать школьные занятия, его освобождают от уроков физкультуры.
Врачи рекомендуют растить и воспитывать ребенка с симптомами наследственного нефрита с учетом безусловного выполнения профилактических мероприятий, которые включают:
- использование только рекомендованного списка продуктов и способа их приготовления;
- нормализацию обменных процессов организма при помощи витаминов, фитотерапии;
- совершение регулярных длительных прогулок.
Содержание:
Синдром Альпорта — генетически детерминированное воспалительное заболевание почек, сопровождающееся поражением слухового и зрительного анализаторов. Это достаточно редкая наследственная патология, встречающаяся у 1 из 10 тысяч новорожденных детей. По данным ВОЗ лица с синдромом Альпорта составляют 1% от всех больных с дисфункцией почек. По МКБ-10 заболевание имеет код Q87.8.
При синдроме Альпорта поражается ген, кодирующий строение белка коллагена, расположенного в базальной мембране почечных канальцев, внутреннего уха и органа зрения. Основная функция базальной мембраны – поддержание и отделение тканей друг от друга. Наследственная неиммунная гломерулопатия проявляется гематурией, нейросенсорной тугоухостью, расстройством зрения. По мере прогрессирования синдрома у больных развивается почечная недостаточность, к которой присоединяются заболевания глаз и ушей. Болезнь носит прогрессирующий характер и не поддается лечению.
Наследственный нефрит или семейный гломерулонефрит — наименования одной и той же патологии. Впервые ее описал в 1927 году ученый из Великобритании Артур Альпорт. Он наблюдал за членами одной семьи, которые страдали тугоухостью и имели эритроциты в анализах мочи. Спустя несколько лет были выявлены поражения глаз у лиц с данным заболеванием. И только в 1985 году ученые установили причину таких аномалий. Ею стала мутация гена, отвечающего за синтез и строение коллагена IV типа.
Чаще всего именно этот недуг становится причиной тяжелой почечной дисфункции у лиц мужского пола. Женщины могут передать мутантный ген своим детям, не имея клинических проявлений. Синдром манифестирует с первых лет жизни. Но чаще всего обнаруживается у малышей в возрасте 3-8 лет. У больных детей сначала появляются признаки поражения почек. Проблемы со слухом и зрением развиваются несколько позже. В позднем детском и юношеском возрасте формируется тяжелая патология почек, потеря зрения и слуха.
По способу наследования аномалии выделяют 3 формы патологии: Х-сцепленная доминантная, аутосомно-рецессивная, аутосомно-доминантная. Каждой форме соответствуют те или иные морфологические и функциональные изменения внутренних органов. В первом случае развивается классическая форма, при которой воспаление почечной ткани проявляется кровью в моче и сопровождается снижение слуха и зрения. При этом болезнь имеет прогрессивное течение, быстро развивается недостаточность почек. Гистологической особенностью подобных процессов является истончение базальной мембраны. Во втором случае врожденный недуг протекает намного легче и отличается изолированным воспалением почек с гематурией. Аутосомно-доминантная форма также считается доброкачественной, отличается благоприятным прогнозом и проявляется только гематурией или же протекает бессимптомно.
Обнаруживают наследственное воспаление почек случайно, во время профосмотра или диагностического обследования других заболеваний.
Этиология
Истинные этиопатогенетические факторы патологии до сих пор полностью не изучены. Предполагают, что синдром Альпорта — наследственное заболевание, обусловленное мутацией гена, расположенного в длинном плече Х хромосомы и кодирующего белок коллаген IV типа. Основная функция коллагена – обеспечение прочности и эластичности соединительнотканных волокон. При данном синдроме отмечается поражение сосудистой стенки почек, кортиева органа, капсулы хрусталика.
Мутантный ген чаще всего передается от родителей детям. Существует основные формы наследования патологии:
- Доминантный Х-сцепленный тип наследования характеризуется передачей пораженного гена от матери сыну или дочери, а от отца – только дочери. Синдром более тяжело протекает у мальчиков. У больных отцов рождаются здоровые сыновья и больные дочери.
- Аутосомно-рецессивный тип характеризуется получением одного гена от отца, а второго — от матери. Больные дети рождаются в 25% случаев, причем одинаково часто как среди девочек, так и среди мальчиков.
В семье с наследственными заболеваниями мочевыделительной системы вероятность рождения больных детей увеличивается в разы. Если больной ребенок рождается в семье, где все члены имеют идеально здоровые почки, причиной синдрома является спонтанная генетическая мутация.
Факторы, способствующие развитию болезни:
- родственники с почечными патологиями;
- родственные браки;
- сдвиги со стороны иммунной системы;
- снижение слуха в молодом возрасте;
- острые инфекции бактериального или вирусного происхождения;
- вакцинация;
- физическое перенапряжение.
Экспрессия мутантного гена у разных индивидуумов варьируется от слабой до значительной выраженности клинических проявлений наследственного нефрита. Процесс разрушения базальной мембраны находится в непосредственной зависимости от тяжести патологического процесса.
Патогенез
Патогенетические звенья синдрома:
- нарушение биосинтеза коллагена или его дефицит,
- деструкция базальной мембраны почек, внутреннего уха и глазного аппарата,
- прорастание коллагеновых волокон V и VI типов,
- поражение почечных клубочков,
- иммунонегативный гломерулит,
- гиалиноз клубочков, атрофия канальцев и фиброз стромы почек,
- гломерулосклероз,
- скопление в почечной ткани липидов и липофагов,
- снижение в крови уровня Ig A, повышение IgM и G,
- снижение активности Т- и В-лимфоцитов,
- нарушение фильтрационной функции почек,
- дисфункция органа зрения и слуха,
- накопление в крови токсинов и продуктов обмена,
- протеинурия,
- гематурия,
- развитие острой почечной недостаточности,
- смерть.
Заболевание развивается постепенно с ренальных симптомов. На ранних стадиях патологии почки работают полноценно, в моче имеются следы белка, лейкоцитов и крови. Поллакиурия и никтурия сопровождаются гипертензией и другими признаками мочевого синдрома. У больных расширяются чашечки и лоханки почек, возникает аминоацидурия. Спустя некоторое время присоединяется тугоухость неврогенного происхождения.
Мужчины в наибольшей степени подвержены развитию почечной дисфункции. При отсутствии лечения смерть наступает в возрасте 15-30 лет. Женщины обычно страдают скрытой формой патологии с признаками гематурического синдрома и незначительным снижением слуха.
Симптоматика
Наследственный нефрит у детей может протекать по гломерулонефротическому или пиелонефротическому типу. Клинические признаки синдрома Альпорта условно делятся две большие группы – ренальные и экстраренальные.
Основными проявлениями почечной симптоматики являются: гематурия – кровь в моче и протеинурия – белок в моче. Эритроциты в моче у больных детей появляются сразу после рождения. Сначала это бессимптомная микрогематурия. Ближе к 5-7 годам кровь в моче становится отчетливо видна. Это патогномоничный признак синдрома Альпорта. Интенсивность гематурии возрастает после острых инфекционных заболеваний — ОРВИ, ветряной оспы, кори. Активные физические нагрузки и профилактические прививки также могут спровоцировать значительное повышение эритроцитов в крови. Несколько реже у мальчиков развивается протеинурия. У девочек этот симптом обычно отсутствует. Потеря белка с мочой сопровождается отеками, повышением артериального давления, общей интоксикацией организма. Возможна лейкоцитурия без бактериурии, анемия.
Развиваясь, болезнь Альпорта осложняется развитием почечной недостаточности. Ее классические признаки — сухая, желтоватая кожа, снижение тургора, сухость во рту, олигурия, тремор кистей, ломота в мышцах и суставах. При отсутствии правильного лечения возникает терминальная стадия патологии. В таких случаях поможет поддержать жизненные силы организма только гемодиализ. Своевременная заместительная терапия или пересадка больной почки позволяют продлить жизнь больным.
К внепочечным симптомам относятся:
- тугоухость, обусловленная невритом слухового нерва;
- ухудшение зрения, связанное с катарактой, изменением формы хрусталика, появлением белых или желтых вкраплений на сетчатке в районе макулы, миопией, кератоконусом;
- задержка в психофизическом развитии;
- врожденные дефекты – высокое небо, синдактилия, эпикант, деформация ушей, патологии прикуса;
- лейомиоматоз пищевода, трахеи, бронхов.
К неспецифическим общеинтоксикационным признакам патологии относятся:
- головная боль,
- миалгия,
- головокружение,
- резкие колебания артериального давления,
- одышка,
- частое, поверхностное дыхание,
- шум в ушах,
- бледность кожи,
- частые позывы к мочеиспусканию,
- диспепсия,
- ухудшение аппетита,
- нарушение режима сна и бодрствования,
- зуд кожи,
- судороги,
- боль в груди,
- спутанность сознания.
У больных развивается компенсированная клубочковая и канальцевая недостаточность, нарушается транспорт аминокислот и электролитов, концентрационная способность почек, ацидогенез, поражается система канальцев нефрона. По мере прогрессирования патологии признаки мочевого синдрома дополняются выраженной интоксикацией, астенизацией и анемизацией организма. Подобные процессы развиваются у мальчиков, имеющих пораженный ген. У девочек заболевание протекает намного легче, стойкая дисфункция почек у них не развивается. Только во время беременности девушки страдают от симптомов недуга.
Осложнения синдрома Альпорта развиваются при отсутствии адекватной терапии. У больных нарастают признаки недостаточности почек: появляются отеки на лице и конечностях, гипотермия, охриплость, олигурия или анурия. Часто присоединяется вторичная бактериальная инфекция – развивается пиелонефрит или гнойный отит. В таком случае прогноз неблагоприятный.
Диагностика
Диагностикой и лечением синдрома Альпорта занимаются педиатры, нефрологи, генетики, ЛОР-врачи, офтальмологи.
Диагностические мероприятия начинаются со сбора анамнеза и выслушивания жалоб больного. Особое значение имеет семейный анамнез. Специалисты выясняют, имелись ли случаи гематурии или протеинурии у родственников, а также случаи смерти от почечной дисфункции. Для постановки диагноза важны данные генеалогического анализа и акушерского анамнеза.
- Специфическое поражение базальной мембраны у больных обнаруживают по результатам биопсии.
- В общем анализе мочи — эритроциты, белок, лейкоциты.
- Генетическое исследование – выявление мутации генов.
- Аудиометрия обнаруживает нарушения слуха.
- Обследование у офтальмолога позволяет выявить врожденную патологию зрения.
- Ультразвуковое исследование почек и мочеточников, магнитно-резонансная томография, рентген и сцинтиграфия — дополнительные диагностические методики.
Лечение
Синдром Альпорта — неизлечимое заболевание. Замедлить развитие почечной недостаточности помогут следующие рекомендации специалистов:
- Рациональное и витаминизированное питание,
- Оптимальные физические нагрузи,
- Частые и длительные прогулки на свежем воздухе,
- Санация очагов хронической инфекции,
- Профилактика инфекционных заболеваний,
- Запрет на стандартные прививки больным детям,
- Фитосбор из крапивы, тысячелистника и черноплодной рябины показан больным детям с гематурией,
- Витаминотерапия и биостимуляторы для улучшения обмена веществ.
Правильное питание заключается в употреблении легкоусвояемых продуктов с достаточным содержанием основных нутриентов. Из рациона больных следует исключить солености и копчености, пряные и острые блюда, алкоголь, продукты с искусственными красителями, стабилизаторами, ароматизаторами. В случае нарушения функций почек необходимо ограничить потребление фосфора и кальция. Подобные рекомендации следует соблюдать больным в течение всей жизни.
Медикаментозная симптоматическая терапия:
- Для устранения гипертензии назначают ингибиторы АПФ – «Каптоприл», «Лизиноприл» и блокаторы рецепторов ангиотензина – «Лориста», «Вазотенз».
- Пиелонефрит развивается в результате присоединения инфекции. В таком случае применяют антибактериальные и противовоспалительные медикаменты.
- Для коррекции нарушений водно-электролитного обмена назначают «Фуросемид», «Верошпирон», внутривенно физраствор, глюкозу, кальция глюконат.
- Анаболические гормоны и железосодержащие препараты показаны для ускоренного образования эритроцитов.
- Иммуномодулирующая терапия – «Левамизол».
- Антигистаминные препараты – «Зиртек», «Цетрин», «Супрастин».
- Комплекс витаминов и лекарств, улучшающих обмен веществ.
Гипербарическая оксигенация оказывает положительный эффект на выраженность гематурии и функционирование почек. При переходе почечной недостаточности в терминальную стадию требуется гемодиализ и пересадка почки. Оперативное вмешательство проводится после достижения больными пятнадцатилетнего возраста. Рецидив заболевания в трансплантате не отмечается. В отдельных случаях возможно развитие нефрита.
Генотерапия синдрома в настоящее время активно разрабатывается. Ее основная цель – предупреждение и замедление ухудшения функционирования почек. Этот перспективный вариант лечения сегодня внедряется в лечебную практику западными медицинскими лабораториями.
Прогноз и профилактика
Синдром Альпорта — наследственное заболевание, предупредить появление которого просто невозможно. Соблюдение всех предписаний врача и ведение здорового образа жизни помогут улучшить общее состояние больных.
Прогноз синдрома считается благоприятным, если у больных обнаруживается гематурия без протеинурии и тугоухости. Почечная недостаточность не развивается также у женщин без поражения слухового анализатора. Даже при наличии стойкой микрогематурии заболевание у них практически не прогрессирует и не ухудшает общего состояния больных.
Наследственный нефрит в сочетании с быстрым развитием почечной недостаточности имеет неблагоприятный прогноз у мальчиков. У них рано развиваются дисфункции почек, глаз и ушей. При отсутствии своевременного и грамотного лечения больные погибают в возрасте 20-30 лет.
Синдром Альпорта – опасное заболевание, которое без оказания квалифицированной медицинской помощи ухудшает качество жизни пациентов и заканчивается их смертью. Чтобы облегчить течение наследственного нефрита, необходимо неукоснительно соблюдать все врачебные рекомендации.
Видео: лекция по синдрому Альпорта
Администратор 04 Октября в 6:51 —> 27470Синдром Альпорта (семейный гломерулонефрит) – это редкое генетическое заболевание, которое характеризуется гломерулонефритом, прогрессирующей почечной недостаточностью, нейросенсорной тугоухостью и поражением глаз. Заболевание было впервые описано британским врачом Артуром Альпортом в 1927 году. Синдром Альпорта встречается очень редко, но в США он отвечает за 3% случаев терминальной почечной недостаточности у детей и 0,2% у взрослых, а также считается наиболее распространенным типом семейного нефрита. Тип наследования синдрома Альпорта может быть разным: • Х-сцепленный доминантный (XLAS): 85%. • Аутосомно-рецессивный (ARAS): 15%. • Аутосомно-доминантный (ADAS): 1%. Наиболее распространенная Х-сцепленная форма синдрома Альпорта приводит к терминальной стадии почечной недостаточности у мужчин. Гематурия обычно возникает у мальчиков с синдромом Альпорта в первые годы жизни. Протеинурия обычно отсутствует в детстве, но это состояние часто развивается у мужчин с XLAS и у представителей обоих полов с ARAS. Потеря слуха и поражение глаз никогда не обнаруживаются при рождении – они возникают в позднем детстве или в юности, незадолго до развития почечной недостаточности.
Причины и механизм развития синдрома Альпорта
Синдром Альпорта вызван мутациями в генах COL4A4, COL4A3, COL4A5, отвечающих за биосинтез коллагена. Мутации в указанных генах нарушают нормальный синтез коллагена типа IV, который является очень важным структурным компонентом базальных мембран в почках, внутреннем ухе и глазах. Базальные мембраны – это тонкие пленочные структуры, которые поддерживают ткани и отделяют их друг от друга. При нарушении синтеза коллагена типа IV гломерулярные базальные мембраны в почках не способны нормально фильтровать токсичные продукты из крови, пропуская в мочу белки (протеинурия) и эритроциты (гематурия). Аномалии синтеза коллагена типа IV приводят к почечной недостаточности и отказу почек, что и является главной причиной смерти при синдроме Альпорта.
Клиника
Гематурия – это наиболее частое и раннее проявление синдрома Альпорта. Микроскопическая гематурия наблюдается у 95% женщин и практически у всех мужчин. У мальчиков гематурию обычно выявляют в первые годы жизни. Если у мальчика за первые 10 лет жизни не обнаружена гематурия, то американские эксперты рекомендуют считать, что у него маловероятно наличие синдрома Альпорта. Протеинурия в детстве обычно отсутствует, но иногда развивается у мальчиков с Х-сцепленным синдромом Альпорта. Протеинурия, как правило, прогрессирует. Значительная протеинурия у больных женского пола встречается нечасто. Гипертензия чаще присутствует у пациентов мужского пола с XLAS и у больных обоих полов с ARAS. Частота и тяжесть гипертензии повышается с возрастом и по мере прогрессирования почечной недостаточности. Нейросенсорная тугоухость (нарушение слуха) – это характерное проявление синдрома Альпорта, которое наблюдается довольно часто, но не всегда. Есть целые семьи с синдромом Альпорта, которые страдают от тяжелой нефропатии, но имеют нормальный слух. Нарушение слуха никогда не обнаруживается при рождении. Билатеральная высокочастотная нейросенсорная тугоухость обычно проявляется в первые годы жизни или в раннем подростковом возрасте. На ранней стадии болезни нарушение слуха определяется только при аудиометрии. По мере прогрессирования, нарушение слуха распространяется на низкие частоты, включая человеческую речь. После появления тугоухости следует ожидать вовлечения почек. Американские ученые утверждают, что при Х-связанном синдроме Альпорта 50% мужчин страдают нейросенсорной тугоухостью к 25 годам, а к 40 годам – около 90%. Передний лентиконус (выпячивание центрального участка хрусталика глаза вперед) наблюдается у 25% пациентов с XLAS. Лентиконуса нет при рождении, но с годами он приводит к прогрессирующему ухудшению зрения, которое заставляет больных часто менять очки. Состояние не сопровождается болью в глазах, покраснением или нарушениями цветового зрения. Ретинопатия – это самое распространенное проявление синдрома Альпорта со стороны органа зрения, поражает 85% мужчин с Х-сцепленной формой болезни. Появление ретинопатии обычно предшествует почечной недостаточности. Задняя полиморфная дистрофия роговицы – редкое состояние при синдроме Альпорта. У большинства нет никаких жалоб. Мутация L1649R в гене коллагена COL4A5 может также вызывать истончение сетчатки, которое ассоциируется с Х-сцепленным синдромом Альпорта. Диффузный лейомиоматоз пищевода и бронхиального дерева – еще одно редкое состояние, которое наблюдается в некоторых семьях с синдромом Альпорта. Симптомы появляются в позднем детском возрасте и включают нарушение глотания (дисфагия), рвоту, боль в эпигастрии и за грудиной, частые бронхиты, одышку, кашель. Лейомиоматоз подтверждается компьютерной томографией или МРТ.
Аутосомно-рецессивная форма синдрома Альпорта
На ARAS приходится всего 10-15% случаев болезни. Эта форма встречается у детей, чьи родители являются носителями одного из пораженных генов, сочетание которых у ребенка вызывает болезнь. Сами родители не имеют симптомов или имеют незначительные проявления, а дети тяжело больны – их симптомы напоминают XLAS.
Аутосомно-доминантная форма синдрома Альпорта
ADAS – это самая редкая форма синдрома, которая затрагивает одно поколение за другим, причем мужчины и женщины болеют одинаково тяжело. Почечные проявления и глухота напоминают XLAS, но почечная недостаточность может возникать в более позднем возрасте. Клинические проявления ADAS дополняются склонностью к кровотечениям, макротромбоцитопенией, синдромом Эпштейна, наличием нейтрофильных включений в крови.
Диагностика синдрома Альпорта
• Лабораторные анализы. Анализ мочи: у больных с синдромом Альпорта чаще всего присутствует кровь в моче (гематурия), а также высокое содержание белка (протеинурия). Анализы крови демонстрирует почечную недостаточность. • Биопсия тканей. Ткань почек, полученную при биопсии, исследуют с помощью электронной микроскопии на наличие ультраструктурных аномалий. Биопсия кожи менее инвазивна, и американские эксперты рекомендуют выполнять ее в первую очередь. • Генетический анализ. В диагностике синдрома Альпорта, если остаются сомнения после биопсии почки, генетический анализ используется для получения однозначного ответа. Определяются мутации генов синтеза коллагена типа IV. • Аудиометрия. Все дети с семейной историей, позволяющей заподозрить синдром Альпорта, должны проходить высокочастотную аудиометрию для подтверждения нейросенсорной тугоухости. Рекомендуется периодический мониторинг. • Обследование глаз. Обследование у офтальмолога очень важно для раннего выявления и мониторинга переднего лентиконуса и других аномалий. • УЗИ почек. На поздних стадиях синдрома Альпорта ультразвуковое исследование почек помогает выявить структурные нарушения. Британские специалисты, основываясь на новых данных (2011) по генетическим мутациям у пациентов с Х-сцепленным синдромом Альпорта, рекомендуют анализ на мутации гена COL4A5, если пациент отвечает хотя бы двум диагностическим критериям по Gregory, и анализ COL4A3 и COL4A4, если мутация COL4A5 не обнаружена или подозревается аутосомное наследование.
Лечение синдрома Альпорта
Синдром Альпорта пока неизлечим. Исследования показали, что ингибиторы АПФ могут уменьшать протеинурию и замедлять прогрессирование почечной недостаточности. Таким образом, использование ИАПФ целесообразно у пациентов с протеинурией, независимо от наличия гипертензии. То же самое касается антагонистов АТII-рецепторов. Оба класса препаратов, судя по всему, помогают уменьшить протеинурию путем снижения внутриклубочкового давления. Более того, ингибирование ангиотензина-II, ростового фактора, отвечающего за гломерулярный склероз, теоретически может замедлять склерозирование. Некоторые исследователи предполагают, что циклоспорин способен уменьшать протеинурию и стабилизировать почечную функцию у пациентов с синдромом Альпорта (исследования были небольшими). Но отчеты говорят, что ответ пациентов на циклоспорин очень вариабельный, и иногда препарат может ускорять интерстициальный фиброз. При почечной недостаточности стандартная терапия включает эритропоэтин для лечения хронической анемии, препараты для контроля остеодистрофии, коррекцию ацидоза и антигипертензивную терапию для контроля АД. Применяется гемодиализ и перитонеальный диализ. Больным с синдромом Альпорта трансплантация почки не противопоказана: опыт пересадки в США показал хорошие результаты. Генная терапия при разных формах синдрома Альпорта является перспективным вариантом лечения, который сегодня активно изучается западными медицинскими лабораториями.Похожие статьиФотогалерея —> БИБЛИОТЕКА МАМЫСиндром Альпорта у детей: особенности развития, симптомы, методы диагностики и лечение21 мая 2019, 13:44Содержание:Синдром Альпорта у детей (иначе – заболевание тонких мембран) – редкая наследственная патология почек, характеризующаяся изменением выработки коллагеновых волокон IV типа, при этом меняется мембранная структура почечных клубочков с одновременным изменением структуры отделов внутреннего уха и глаз. По статистике, встречается 1 случай на 5000 человек. Примечательно, что синдром диагностируют у 1% больных с различными нефроурологическими патологиями и у 3% пациентов, ранее перенесших трансплантацию почки.
Причины и пути наследования
Механизм развития заболевания обусловлен дефектностью генетической единицы, отвечающей за структуру цепей коллагена IV типа. По типу наследования синдром классифицируют:
- на доминантный, или X-сцепленный. Заболевание обусловлено мутацией гена в женской половой хромосоме X. Такой путь наследования встречается в 85% всех клинических случаев;
- аутосомно-рецессивный. Обусловлен мутациями в генах второй порядковой хромосомной единицы, встречается в 10% всех случаев синдрома Альпорта;
- аутосомно-доминантный. Наследственный нефрит вызван мутацией генов во второй порядковой хромосоме, встречается в 1–2% случаев и напоминает течение аутосомно-рецессивного типа.
По мере прогрессирования болезни полностью разрушаются оболочки гломерул, почечных канальцев, внутренних глазных и ушных структур. Механизм развития патологического изменения почечных структур можно объяснить несколькими ключевыми факторами: генной мутацией, нарушением выработки и строения коллагена, разрушением канальцевых и гломерулярных почечных единиц, патологией почек.
Типичные признаки
Болезнь тонких базальных мембран редко диагностируется сразу после рождения или внутриутробно. Специфические симптомы наблюдаются спустя 3–4 года после рождения и выражаются в следующих состояниях:
- гематурический синдром. Одно из ранних проявлений болезни. Обычно выявляется лабораторно. У мальчиков микрогематурия отмечается еще до 1 года, но планово такой анализ на фоне общего здоровья не проводится. Ученые считают, что если до 10 лет ребенка гематурия не выявлялась ни разу, то синдром Альпорта можно исключать на 98%;
- белок в моче. Состояние протеинурии чаще отмечается у мальчиков с доминантным путем наследования мутации. Обычно белок в моче не выявляется или его появление носит эпизодический характер;
- артериальная гипертензия. Стойкое повышение артериального давления характеризуется прогрессированием почечной недостаточности. Чем ниже клиренс креатинина, тем интенсивнее развитие гипертензии;
- тугоухость. Нарушение слуха на фоне нефротического синдрома возникает не всегда. Известны случаи семейного синдрома Альпорта, когда у всех больных нет слуховых изменений. Ранняя стадия тугоухости определяется только при аудиометрии;
- лентиконус передний. Состояние характеризуется выпячиванием глазного хрусталика кнаружи, встречается у 20–25% больных с наследственным нефритом. Офтальмологическое заболевание постоянно прогрессирует, поэтому больные вынуждены часто менять очки;
- роговичная дистрофия. Истончение сетчатки глаза встречается редко. Почти 90% больных с дистрофией роговицы имеют X-сцепленный путь геномной мутации;
- лейомиоматоз бронхов и пищевода. У детей возникает ранняя дисфагия – нарушение глотания. Появляются и иные симптомы: загрудинные и эпигастральные боли, рвота, одышка, сухой кашель. Подтвердить лейомиоматоз можно с помощью МРТ или компьютерной томографии.
По мере взросления ребенка нарастают симптомы почечной недостаточности: недомогание, интоксикация, артериальная гипертензия, резкое увеличение креатинина и мочевины в составе крови, ацидоз.
Диагностика
Комплексное обследование детей с синдромом Альпорта включает сбор анамнеза жизни, наследственного и клинического анамнеза, изучение жалоб. Учитывая наследственную природу заболевания, значение имеют случаи почечной недостаточности у близких родственников. Для уточнения диагноза проводят ряд исследований:
- общеклинические лабораторные исследования, в том числе на синдром Альпорта;
- УЗИ почек и почечных структур, органов мочеполовой системы;
- рентгенконтрастные методы исследования (экскреторная урография);
- ДНК-тест;
- биопсия почек.
Диагностика требует дополнительной консультации нефролога, уролога, генетика, офтальмолога, кардиолога, отоларинголога. Заболевание дифференцируют от других геномных мутаций, в частности, от синдрома «кошачьего крика» — нарушения строения 5 хромосомы, когда типичные признаки выражаются в нарушении слуха и зрения еще в раннем возрасте.
Лечение
Заболевание неизлечимо, на причины генетических мутаций невозможно повлиять медикаментозно или хирургически. Терапия направлена на облегчение симптомов болезни. Обычно схема лечения включает следующие группы препаратов:
- мочегонные (петлевые, тиазидные, растительного происхождения);
- внутривенное введение физраствора для предупреждения обезвоживания;
- средства на основе глюконата кальция и глюкозы для восстановления минерального и электролитного баланса;
- антигипертензивная терапия при вторичной артериальной гипертензии (ингибиторы АПФ, блокаторы медленных кальциевых каналов);
- гормоны;
- железосодержащие препараты при выраженном гематурическом синдроме.
При развитии тяжелой почечной недостаточности встает вопрос о заместительной терапии. Лучшим способом заместительной терапии у детей является трансплантация почки. Пересадка органа позволяет вернуть прежний образ жизни, улучшить состояние больного.
Общие клинические рекомендации для больных наследственной нефропатией заключаются в соблюдении охранительного режима и диеты, регулярных прогулках на свежем воздухе, приеме витаминных комплексов. Все лечебные меры должны отвечать возрастным интересам ребенка.
Осложнения
Основными осложнениями при синдроме Альпорта у детей являются глухота, нарушение зрения и недостаточность функции почек. Последнее состояние провоцирует развитие вторичных осложнений, угрожает жизни пациентов.
Прогноз при синдроме Альпорта у детей преимущественно благоприятный, если прогрессия крайне медленная, а симптомы заболевания не выражены. Если гематурия не сопровождается протеинурией, состояние пациента не осложнено, то важно соблюдать предупредительные лечебные меры. Учитывая генетическую природу заболевания, предупредить развитие патологии у детей невозможно.
Также интересно почитать: причины микроцефалии у детей
Гломерулонефрит у детей: общие характеристики, симптомы, лечение и прогноз ›Формы и методы лечения гипоспадии у мальчиков ›Подпишись на канал baby.ru в—>Родителям о детях » Детское здоровье » Болезни по темам » Нефрит
Генетически детерминированная неиммунная гломерулопатия, протекающая с гематурией, прогрессирующим снижением почечных функций — это синдром Альпорта или наследственный нефрит. Он проявляется комплексом патологий: наличием нефрита с гематурией, ухудшением слуха и патологией зрения. В этой статье мы расскажем вам про основные причины и симптомы синдрома, а также о том, как проводится его лечение у ребенка.
<center> </center>
</center>
Причины синдрома Альпорта у детей
Согласно данным эпидемиологических исследований, проведенных в 13 регионах России, это заболевание встречается с частотой 17 на 100 000 детского населения [Игнатова М. С, 1999].
Этиология синдрома Альпорта
Генетическая основа заболевания – мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для базальных мембран почки, кохлеарного аппарата, капсулы хрусталика, сетчатки и роговицы глаза, что доказано в исследованиях с использованием моноклональных антител против этой фракции коллагена. В последнее время указывают на возможность применения ДНК-зондов для пренатальной диагностики заболевания [Цаликова Ф. Д. и др., 1995].
Подчеркивается важность тестирования всех членов семьи с помощью ДНК-зондов для выявления носителей мутантного гена, что имеет большое значение при проведении медико-генетического консультирования семей с этим заболеванием. Однако до 20% семей не имеют родственников, страдающих болезнью почек, что позволяет предполагать высокую частоту спонтанных мутаций аномального гена.
У большинства больных синдромом Альпорта, в семьях имеются лица с почечными заболеваниями, снижением слуха и патологией зрения, имеют значение родственные браки между людьми, имеющими одного или более предков, т.к. в браке родственных особей возрастает вероятность получения одинаковых генов со стороны обоих родителей [Фокеева В. В. и др., 1988]. Установлены аутосомно-доминантный и аутосомно-рецессивный и доминантный, сцепленный с Х-хромосомой пути передачи.
У малышей чаще различают три варианта синдрома Альпорта:
- непосредственно сам синдром,
- наследственный нефрит без тугоухости,
- семейная доброкачественная гематурия.
Патогенез синдрома Альпорта
В основе лежит сочетанный дефект структуры колагена базальной мембраны клубочков почек, структур уха и глаза. Ген классического синдрома расположен в локусе 21-22 q длинного плеча Х-хромосомы. В большинстве случаев наследуется по доминантному типу, сцепленному с Х-хромосомой. В связи с этим у мужчин синдром Альпорта протекает тяжелее, так как у женщин функция мутантного гена компенсируется здоровым аллелем второй, неповрежденной хромосомы.
При исследовании биоптата почек по данным электронной микроскопии наблюдаются такие симптомы: ультраструктурные изменения базальной мембраны клубочка: истончение, нарушение структуры и расщепление гломерулярных базальных мембран с изменением ее толщины и неравномерностью контуров. На ранних стадиях заболевания дефект определяет истончение и ломкость гломерулярных базальных мембран.
Истончение гломерулярных мембран является более благоприятным признаком и чаще встречается у девочек. Более постоянный электронно-микроскопический признак при данной болезни – расщепление базальной мембраны, причем выраженность деструкции ее коррелирует с тяжестью процесса.
<center> </center>
</center>
По каким симптомам узнать синдром Альпорта у детей?
Первые симптомы заболевания в виде изолированного мочевого синдрома чаще выявляются у малышей первых трех лет жизни. В большинстве случаев заболевание обнаруживается случайно. Мочевой синдром выявляется при профилактическом обследовании ребенка, перед поступлением в детское учреждение или во время ОРВИ. В случае появления патологии в моче во время ОРВИ при синдроме в отличие от приобретенного гломерулонефрита отсутствует латентный период.
Как проявляется синдром Альпорта в начальной стадии?
В начальной стадии самочувствие ребенка страдает мало, симптомы не явно выражены, проводится лечение согласно рекомендациям врача. Характерной особенностью является упорство и стойкость мочевого синдрома. Одним из основных признаков является гематурия различной степени выраженности, наблюдаемая в 100% случаев. Усиление степени гематурии отмечается во время или после инфекций дыхательных путей, физической нагрузки или после профилактических прививок. Протеинурия в большинстве случаев не превышает 1 г/сут, в начале заболевания может быть непостоянной, по мере прогрессирования процесса протеинурия нарастает. Периодически в мочевом осадке может присутствовать лейкоцитурия с преобладанием лимфоцитов, что связывают с развитием интерстициальных изменений.
В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного: появляются интоксикация, мышечная слабость, артериальная гипотония, часто нарушение слуха (особенно у мальчиков), иногда нарушение зрения. Интоксикация проявляется бледностью, утомляемостью, головными болями.
Снижение слуха — симптом появления синдрома Альпорта
В начальной стадии болезни снижение слуха в большинстве случаев выявляется только с помощью аудиографии. Снижение слуха может возникнуть в различные периоды детства, однако чаще всего тугоухость диагностируется в возрасте 6 — 10 лет. Начинается с высоких частот, достигая значительной степени при воздушном и костном проведении, переходя из звукопроводящей в звуковоспринимающую тугоухость. Снижение слуха может быть одним из первых симптомов заболевания и может предшествовать мочевому синдрому.
Снижение зрения — симптом синдрома Альпорта
В 20% случаев у больных отмечаются изменения со стороны органов зрения. Наиболее часто выявляются аномалии со стороны хрусталика: сферофокия, лентиконус передний, задний или смешанный, разнообразные катаракты. В семьях с с этим заболеванием наблюдается значительная частота миопии. Ряд исследователей постоянно в этих семьях отмечают билатеральные перимакулярные изменения в виде ярких беловатых или желтоватых грануляций в области желтого тела. Они считают этот признак постоянным симптомом, который имеет высокую диагностическую ценность при этом заболевании. К. S. Chugh и соавт. (1993) при офтальмологическом исследовании выявили у больных снижение остроты зрения в 66,7% случаев, передний лентиконус – в 37,8%, пятна на сетчатке – в 22,2%, катаракту – в 20%, кератоконус – в 6,7%.
Особенности синдрома Альпорта у детей
У части детей, особенно при формировании почечной недостаточности, отмечают существенное отставание в физическом развитии. По мере прогрессирования почечной недостаточности развивается артериальная гипертензия. У ребенка ее симптомы чаще выявляются в подростковом периоде и в более старших возрастных группах. При диагностировании лечение проводят незамедлительно.
Характерно наличие у больных с синдромом Альпорта разнообразных (более 5 — 7) стигм соединительнотканного дизэмбриогенеза [Фокеева В. В., 1989]. Среди соединительнотканных стигм у больных наиболее часто встречаются гипертелоризм глаз, высокое нёбо, аномалии прикуса, аномальная форма ушных раковин, искривление мизинца на руках, «сандалевидная щель» на стопах. Для заболевания характерны такие симптомы: однотипность стигм дизэмбриогенеза внутри семьи, а также высокая частота их распространения у родственников пробандов, по линии которых передается болезнь.
Клинические рекомендации о синдроме Альпорта
На ранних этапах болезни выявляется изолированное снижение парциальных функций почек: транспорта аминокислот, электролитов, концентрационной функции, ацидогенеза, в дальнейшем изменения касаются функционального состояния как проксимального, так и дистального отдела нефрона и носят характер сочетанных парциальных нарушений. Снижение клубочковой фильтрации наступает позднее, чаще в подростковом периоде. По мере прогрессирования развивается анемизация.
Таким образом, характерна стадийность течения заболевания: сначала латентная стадия или скрытых клинических симптомов, проявляющаяся минимальными изменениями мочевого синдрома, затем наступает постепенная декомпенсация процесса со снижением почечных функций с манифестными клиническими симптомами (интоксикация, астенизация, отставание в развитии, анемизация). Клинические симптомы появляются обычно вне зависимости от наслоения воспалительной реакции.
Синдром может манифестовать в разные возрастные периоды, что зависит от действия гена, который до определенного времени находится в репрессированном состоянии.
Как диагностируют синдром Альпорта у детей?
Предложены следующие критерии:
- Наличие в каждой семье не менее двух больных нефропатией,
- Гематурия как ведущий симптом нефропатии у пробанда,
- Наличие тугоухости хотя бы у одного из членов семьи,
- Развитие ХПН у одного родственника и более.
При диагностике разнообразных наследственных и врожденных заболеваний большое место принадлежит комплексному подходу к обследованию и прежде всего обращение внимания на данные, получаемые при составлении родословной ребенка. Диагноз синдрома Альпорта считается правомочным в случаях обнаружения у больного 3 из 4 типичных признаков: наличие в семье гематурии и хронической почечной недостаточности, присутствие у больного нейросенсорной тугоухости, патологии зрения, обнаружение при электронно-микроскопической характеристике биоптата признаков расщепления гломерулярной базальной мембраны с изменением ее толщины и неравномерностью контуров [Игнатова М. С, 1996].
Клинико-генетические методы исследования синдрома Альпорта
Прежде чем начнется лечение, проводится обследование больного, которое должно включать клинико-генетические методы исследования, направленное изучение анамнеза заболевания, общий осмотр больного с учетом диагностически значимых критериев.
- В стадии компенсации уловить патологию можно лишь ориентируясь на такие синдромы, как наличие наследственной отягощенности, гипотонии, множественных стигм дизэмбриогенеза, изменения мочевого синдрома.
- В стадии декомпенсации возможно появление эстраренальных симптомов, таких как выраженная интоксикация, астенизация, отставание в физическом развитии, анемизация, проявляющиеся и усиливающиеся с постепенным снижением почечных функций. У большинства больных при снижении почечных функций наблюдается: снижение функции ацидо- и аминогенеза, у 50% больных отмечают значительное снижение секреторной функции почек, ограничение пределов колебания оптической плотности мочи, нарушение ритма фильтрации, а затем и снижение клубочковой фильтрации.
- Стадия хронической почечной недостаточности диагностируется при наличии у больных в течение 3-6 мес. и более повышенного уровня мочевины в сыворотке крови (более 0,35 г/л), снижение клубочковой фильтрации до 25% от нормы.
Дифференциальная диагностика синдрома Альпорта
Ее приходится проводить с гематурической формой приобретенного гломерулонефрита. Приобретенный гломерулонефрит имеет чаще острое начало, период 2 — 3 недели после перенесенной инфекции, экстраренальные признаки, в том числе гипертензию с первых дней (при синдроме Альпорта, напротив, гипотония), снижение клубочковой фильтрации в начале заболевания, отсутствие нарушения парциальных канальцевых функций, тогда как при наследственном они присутствуют. Приобретенный гломерулонефрит протекает с более выраженной гематурией и протеинурией, с увеличенной СОЭ. Диагностическое значение имеют типичные изменения гломерулярной базальной мембраны, свойственные синдрому. Лечение должно быть начато оперативно.
Дифференциальная диагностика от дисметаболической нефропатии проводится с хронической почечной недостаточностью, в семье клинически выявляются неоднотипные болезни почек, а может быть спектр нефропатии от пиелонефрита до мочекаменной болезни. Часто присутствуют жалобы на боли в животе и периодически при мочеиспускании, в осадке мочи – оксалаты.
При подозрении на заболевание, больного необходимо направить для уточнения диагноза в специализированное нефрологическое отделение.
<center> </center>
</center>
Как лечить синдром Альпорта у детей?
В режиме лечения предусматривается ограничение от больших физических нагрузок, пребывание на свежем воздухе. В период, когда проводится лечение показана полноценная диета, с достаточным содержанием полноценных белков, жиров и углеводов с учетом функции почек. Большое значение имеет выявление и санация хронических очагов инфекции. Из лекарственных средств используются АТФ, кокарбоксилаза, пиридоксин (до 50 мг/сут.), витамин В5, карнитина хлорид. Курсы проводят 2 — 3 раза в год. При гематурии назначается фитотерапия – крапива двудомная, сок черноплодной рябины, тысячелистник.
В зарубежной и отечественной литературе имеются сообщения о лечении преднизолоном и использовании цитостатиков. Однако об эффекте судить трудно.
Лечение синдрома Альпорта
При хронической почечной недостаточности применяются гемодиализ и трансплантация почек.
М. С. Игнатова (1999) считает, что основным методом при развитии ХПН является своевременное проведение трансплантации почки, которое возможно и без предварительного экстракорпорального диализа. Актуальной является проблема использования методов генной инженерии.
Необходима преемственность в постоянном наблюдении за больными и передача детей педиатром непосредственно терапевту-нефрологу. Диспансерное наблюдение осуществляется на протяжении всей жизни больного.
Теперь вы знаете основные симптомы и способы лечения синдрома Альпорта у детей. Здоровья вашему малышу!
<center>
Другие статьи по этой теме:
</center>
Ваши отзывы и комментарии
Поля отмеченные * обязательны. HTML тэги отключены.Используемые источники:
- https://prosindrom.ru/genetic/sindrom-alporta.html
- https://sindrom.info/alporta/
- https://medbe.ru/news/meditsina/sindrom-alporta-prichiny-i-simptomy-diagnostika-i-lechenie-sindroma-alporta/
- https://m.baby.ru/wiki/sindrom-al-porta-u-detej-osobennosti-razvitia-simptomy-metody-diagnostiki-i-lecenie/
- https://www.medmoon.ru/rebenok/sindrom_alporta_u_detei.html






