
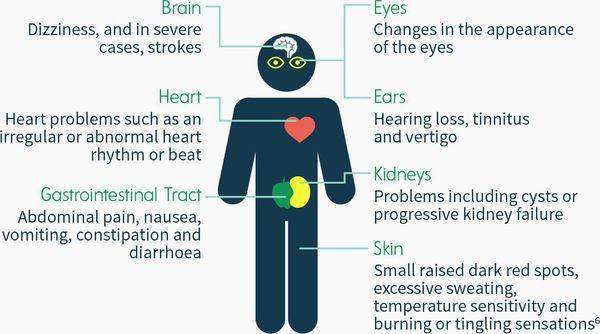
Подробная характеристика клинических симптомов
Типичными для данного заболевания являются мучительные приступы акропарестезии, при которых больной ощущает чувство жжения, покалывания, дискомфорта в дистальных отделах конечностей, возникающие при небольшом болевом раздражении. Тяжесть таких приступов с годами значительно возрастает, приступы случаются чаще и становятся продолжительнее. Иногда приступы такой боли длятся по несколько дней, сопровождаются субфебриллитетом, признаками воспаления по анализу крови.[4] Для болезни Андерсона-Фабри характерно наличие болевых кризов — возникающих периодически интенсивных болевых ощущений, которые воспринимаются как жжение, покалывание, онемение, локализуются в дистальных отделах конечностей, часто распространяются на проксимальные отделы конечностей, туловище. Спровоцировать ухудшение состояния могут стрессовые состояния, перегрев, переохлаждение, изменения атмосферного давления, физическая нагрузка, усталость.[2]
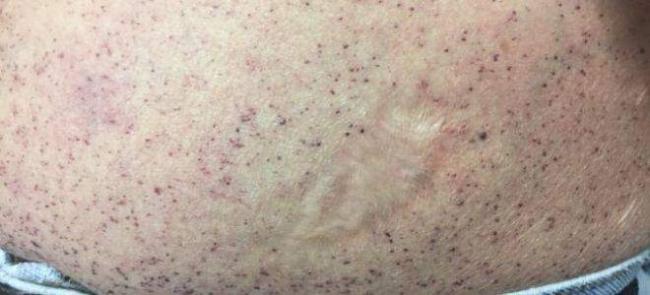
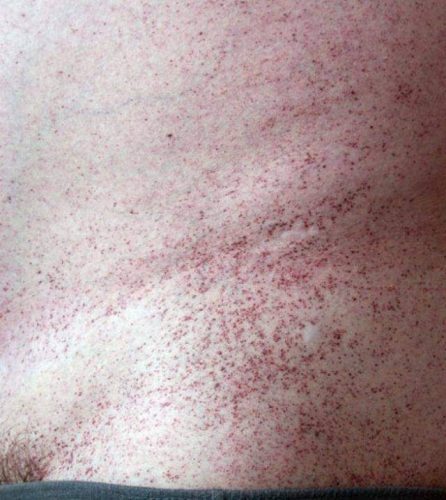
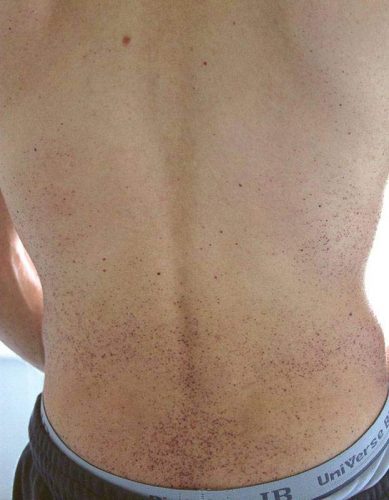
Ангиокератома является типичным поражением сосудов при болезни Фабри. Состоит из конгломерата нескольких расширенных сосудов, покрытых поверхностными слоями кожи. Локализуется на пальцах верхних и нижних конечностей, области вокруг губ, в районе гениталий, ануса, области вокруг пупка, на коленях. Не изменяет цвета при давлении. Представляет из себя пятна вишневого цвета, слегка приподнятые над поверхностью кожи, безболезненные. С годами возрастает их количество и размер. Ангиокератома обычно манифестирует в раннем возрасте и прогрессирует со временем.
Возможно развитие ангиэктазий на слизистых оболочках полости рта и глаз.[2]
К офтальмологическим симптомам относят увеличение диаметра и извилистый ход сосудов сетчатки и конъюнктивы, помутнение роговицы. При осмотре больных при помощи щелевой лампы иногда визуализируются светлые вихреподобные отложения субстрата в роговице, развивается воронкообразная кератопатия. В начальных стадиях заболевания острота зрения остается неизменной, но со временем развивается значительное помутнение роговицы, что может приводить к слепоте. Поражение зрительного нерва происходит редко, но несет за собой тяжелые последствия.
У пациентов с болезнью Андерсона-Фабри наблюдается нарушение потоотделения по типу гипогидроза (пониженного потоотделения), а иногда и ангидроза (отсутствия потоотделения). Это может приводить к непереносимости высоких температур, явлению перегрева организма.
Многие больные, особенно мужчины, жалуются на плохую переносимость физических нагрузок, утомляемость, что связано с быстрым перегревом, усилением интенсивности парестезий.[1]
С возрастом в патологический процесс вовлекается сердечно-сосудистая система. Депонирование сфинголипидов в кардиомиоцитах и эндотелии сосудов нарушает их функции, приводит к развитию фиброза. Проявляется это чувством одышки, стенокардитическими болями, аритмиями, тахикардией, гипертрофией левого желудочка, выявляемой по УЗИ, МРТ и ЭКГ. Это является наиболее неблагоприятным в прогностическом плане проявлением болезни.
Поражение почек выявляется у всех гемизигот. Прогрессирует ХПН, которая на начальных стадиях имеет скрытое течение, нет повышения артериального давления, выявляется нормальный или слегка повышенный уровень креатинина в сыворотке крови, небольшая протеинурия. Со временем почечная недостаточность достигает терминальной стадии, возникает тяжелая уремия, артериальная гипертензия.
Одними из самых явных проявлений заболевания являются неврологические нарушения. Больные предъявляют жалобы на приступы головокружения, головные боли, потерю сознания. Из-за депонирования сфинголипидов в лизосомах нервных клеток и эндотелии сосудов возникают ишемические состояния головного мозга, в результате чего у больных случаются транзиторные ишемические атаки, ишемические и геморрагические инсульты. Зачастую ишемический инсульт в молодом возрасте является единственным проявлением заболевания, особенно у женщин-гетерозигот и больных с атипичным типом заболевания.[3]
В результате ишемических изменений головного мозга больные становятся забывчивыми, рассеянными, неряшливыми, страдают когнитивные функции.
Больные могут жаловаться на шум, звон в ушах, резкое снижение остроты слуха, доходящей до глухоты. Наблюдается нейросенсорная тугоухость.[4]
Поражение желудочно-кишечного тракта проявляется болью в области живота, диспепсией, поносами, метеоризмом, запорами. У большинства больных эпизоды диареи сменяются запорами, развивается геморрой. Иногда развиваются желудочно-кишечные кровотечения.[1]
У части пациентов наблюдается легкая железодефицитная анемия, не требующая коррекции.[1]
У мужчин отмечается отставание физического и полового развития.
Зачастую пациентам с болезнью Андерсона-Фабри ставят диагноз «ревматическая лихорадка», что связано с эпизодами субфебриллитета и повреждением костной системы, которое проявляется вовлечением в патологический процесс дистальных межфаланговых суставов, снижением минеральной плотности позвонков и асептическими некрозами головок бедренной и таранной костей.
При тяжёлом течении заболевания, мучительных парестезиях нарушается психо-эмоциональная сфера человека, что приводит к депрессивным, тревожным состояниям. В некоторых случаях при интенсивных болевых проявлениях болезни пациенты имеют склонность к суициду.
Патогенез болезни Фабри
Болезнь Фабри наследуется по Х-сцепленному рецессивному типу наследования с полной пенетрантностью и различной экспрессивностью у мужчин (гемизигот).[1] Хотя некоторые авторы предполагают Х-сцепленный доминантный тип с неполной пенетрантностью. Тип наследования в настоящее время является темой для дискуссии у специалистов.[2]
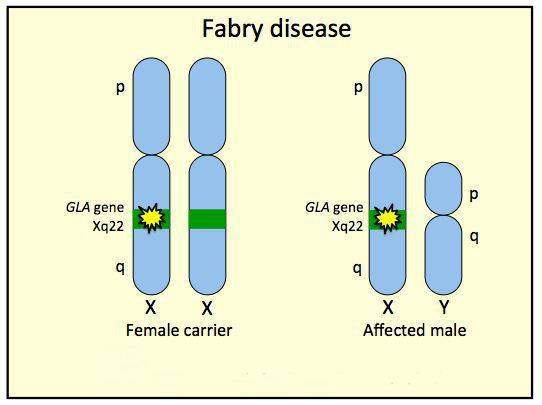
У мужчин-гемизигот заболевание протекает с более выраженными симптомами, как и у женщин-гомозигот. У женщин-гетерозигот зачастую обнаруживается атипичная форма болезни, либо они оказываются здоровыми.[3]
Заболевание вызывается мутацией в гене GLA, локализованном в коротком плече Х-хромосомы (Хq22). Этот ген кодирует синтез фермента альфа-галактозидазы А, ответственного за расщепление сфинголипида глоботриаозилцерамида. Глоботриаозилцерамид в своей структуре несет терминальный остаток альфа-галактозила, для отщепления которого требуется фермент альфа-галактозидаза А. При дефекте гена, кодирующего этот фермент, происходит нарушение этого процесса, что сопровождается отложением гликосфинголипидов с терминальным α-галактозил остатком в клетках.
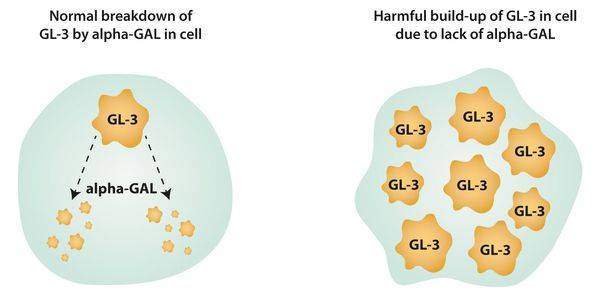
Сфинголипиды — это липиды, производные алифатических аминоспиртов. В случае, когда их разрушение нарушается, происходит избыточное накопление церамидтригексозида в лизосомах клеток.
Накопление субстрата в клетках значительно нарушает их жизнедеятельность, что приводит к изменению функций и клиническим проявлениям заболевания, нарушается нервная передача, связь между клетками.
В сосудистой стенке, почках, миокарде накапливаются сфинголипиды, индуцирующие в последующем процессы фиброгенеза, итогом которых является органная недостаточность.[2]
При накоплении субстрата в эндотелиальной ткани сосудов происходит их сужение, наблюдается нарушение микроциркуляции, гипоксия тканей. Это основной механизм развития ишемических нарушений.
Накопление сфинголипида в нервных клетках приводит к их структурным нарушениям, проявляющимся увеличением числа кальциевых каналов и образованием патологических ноцицептивных связей, повышению возбудимости путей болевой чувствительности, и развитию типичной симптоматики.[2]
Развитие гипогидроза, как и ангидроза, связано с депонированем сфинголипидов в клетках потовых желез, нарушенной иннервацией и уменьшенным кровоснабжением кожи.[2]
Накопление тригексозилцерамида в миокарде вызывает развитие фиброза,[8] прогрессирующей гипертрофической кардиомиопатии, а при наличии сужения коронарных сосудов, это приводит к ишемическим кардиальным симптомам, острому коронарному синдрому, развитию сердечной недостаточности.
Предполагается, что усиление боли при физических нагрузках связано со спазмом суженных сосудов, приводящему к ухудшению трофики нервных волокон.[2]
Отложение субстрата в роговице приводит к развитию ее помутнения, а поражение сосудов может способствовать ухудшению зрения, вплоть до слепоты.
Классификация и стадии развития болезни Фабри
Выделяют две основные формы болезни Фабри:[2]
- классическую;
- атипичную.
Классическая характеризуется ранней манифестацией заболевания (в первом десятилетии жизни), наличием характерных симптомов и осложнений.
При атипичной форме происходит изолированное поражение головного мозга (ранние инсульты), сердца и почек, она манифестирует в более позднем возрасте и представляет большие трудности для диагностики.
Некоторые англоязычные авторы выделяют ещё женскую форму заболевания. Она имеет более лёгкие проявления, начало в среднем на 5-10 лет позднее классической «мужской» формы.
Осложнения болезни Фабри
Со стороны нервной системы:
- ишемические инсульты в молодом возрасте, нередко являющиеся причиной смерти.
Со стороны сердечно-сосудистой системы:
- ишемические инфаркты;
- кардиомиопатии;
- аритмии;
- сердечная недостаточность, приводящая к смерти.
Со стороны почек:
- терминальная стадия почечной недостаточности, требующая трансплантации почек.
Другие системы:
- нарушение зрения; слуха, вплоть до глухоты;
- переломы костей;
- перегрев организма.
Смерть чаще всего наступает от уремии или ишемических поражений мозга и сердца на четвертом десятилетии жизни.[8]
Диагностика болезни Фабри
Ранняя диагностика очень важна для правильного и своевременного назначения лечения пораженных болезнью органов, предотвращения осложнений.[5]
Важным методом диагностики болезни Фабри является оценка генеалогического анамнеза пациента, так как при этом могут обнаружиться родственники со сходными симптомами, которые не знают о своем заболевании. Ген, ответственный за болезнь Андерсона-Фабри, может передаваться через большое число поколений, поэтому больными оказываются многие ближние и дальние родственники. Для определения риска наследования этого заболевания необходимо собрать информацию о здоровье всех известных членов семьи.[3]
Болезнь Фабри бывает очень трудно отличить от более распространенных заболеваний, и пациенты в течение долгих лет могут оставаться без верного диагноза.
Предварительный диагноз ставится по результатам опроса, жалоб пациента, генеалогическому анамнезу.
Для верификации диагноза используют методы обнаружения субстратов и энзимов, ДНК-диагностику.
- Измерение активности альфа-галактозидазы А.[2] При болезни Андерсона-Фабри активность этого фермента у представителей мужского пола всегда ниже нормы, а у женщин этот показатель бывает в пределах нормы или слегка снижен. Материалом для проведения исследования могут служить лейкоциты, плазма крови, слезная жидкость, культура фибробластов.
- Наиболее точным методом диагностики является секвенирование гена GLA.[2] На сегодняшний день описано более пятисот мутаций этого гена, приводящих к болезни Фабри. Использование данной методики ограничено её стоимостью. ДНК-диагностику целесообразно применять для женщин, т. к. у них определение активности альфа-галактозидазы А не всегда дает достоверный результат, и для родственников больного, т. к. они могут являться носителями мутантного гена или больны атипичной формой заболевания. Проведение данного анализа возможно в Центре молекулярной генетики в Москве.
- Количественное определение глоботриаозилцерамида. Этот метод зарекомендовал себя для наблюдения за состоянием пациентов и оценки эффективности лечения, для определения формы заболевания (типичная, атипичная). Материалом для исследования могут быть плазма крови или сухие пятна крови.
- В некоторых случаях проводится биопсия почки с целью обнаружения клеток, содержащих лизосомы с характерным субстратом.
Дифференциальная диагностика болезни Фабри проводится с наследственной геморрагической телеангиэктазией, ревматической лихорадкой, болезнью Фордайса, Шиндлера, другими наследственными болезнями накопления.[1]
Лечение болезни Фабри
Лечение болезни Фабри состоит в замещении дефицитного фермента с помощью ферментозаместительной терапии. Она проводится с помощью внутривенного вливания препарата. Обычно ферментозаместительная терапия используется вместе с различными методами лечения конкретных симптомов.[5]
В России сегодня используются два препарата для ферментозаместительной терапии: Фабразим, Джензайм; Реплагал, Шайер. Эти препараты имеют очень большую стоимость, лечение больных оплачивается из средств федерального бюджета. Так как заболевание относится к орфанным (редким), существует закон об ускоренной процедуре исследования лекарственных препаратов, предназначенных для лечения таких болезней.[3]
Мужчинам начало ферментозаместительного лечения рекомендуется в ближайшее время после установления диагноза.[5]
Для купирования боли применяются препараты из групп атиконвульсантов, антидепрессантов, анальгетиков, нестероидных противовоспалительных препаратов, наркотических анальгетиков.[2]
Больным показано применение антигипертензивных препаратов.
При развитии терминальной почечной недостаточности проводят трансплантацию почки. Донорами не рекомендуется быть женщинам, которые являются родственницами больного.[2]
В качестве коррекции тугоухости показано использование слуховых аппаратов.
Физическая активность пациентов с болезнью Фабри должна быть ограничена из-за возможного усиления симптомов и перегрева организма.[3]
Ситуации, при которых нецелесообразно назначать ферментозаместительную терапию:
- период беременности и лактации;
- если имеется другое опасное для жизни заболевание, при котором прогноз от применения заместительной терапии не станет лучше;
- есть серьезные осложнения (ишемический инсульт, реанимационные больные).
Прогноз. Профилактика
При надлежащем лечении и вовремя начатой заместительной ферментативной терапии прогноз для больных благоприятный. При отсутствии лечения смерть от сердечно-сосудистых, неврологических и почечных осложнений наступает на четвертом десятилетии жизни.[4]
Профилактика заболевания заключается в пренатальной или предимплантационной (при ЭКО) диагностике наличия мутантного гена методом ДНК-диагностики и прерывания беременности по медицинским показаниям.[4] Необходимо обследование всех потенциальных носителей гена для своевременного назначения заместительной ферментативной терапии и предотвращения клинических проявлений болезни и ее осложнений. Возможно скрининговое исследование сухих пятен крови с определением активности альфа-галактозидазы, но оно ограничено финансовыми возможностями региона. Необходимо, чтобы все пациенты с признаками болезни Фабри были исследованы на это заболевание. Для этого существует программа, регламентирующая забор и отправку материала в Центр молекулярной генетики, обеспечение больных препаратами заместительной терапии.
Список литературы
- Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. М: Т-во научных изданий КМК; Авторская академия, 2007. 448 с.
- Федеральные клинические рекомендации по диагностике и лечению болезни Фабри. Москва, 2013.
- Информация о болезни Фабри (дата обращения 25.01.18г.)
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. European Journal of Clinical Investigation 34 (3): 236–42
- Союз пациентов, страдающих болезнью Фабри ((дата обращения 25.01.18г)
- Omid Motabar, Ellen Sidransky, Ehud Goldin, and Wei Zheng. Fabry Disease – Current Treatment and New Drug Development. Curr Chem Genomics. 2010; 4: 50–56
- Stephen Waldek, Sandro Feriozzi. Fabry nephropathy: a review – how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014; 15: 72
- Frank Weidemann, Maria D Sanchez-Niño et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis. 2013; 8: 116
- Juan M. Politei, Didier Bouhassira et al. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci Ther. 2016 Jul; 22(7): 568–576
Клинические cлучаи «Болезнь Фабри»
1.Случай диагностики болезни Фабри у пациента, доставленного скорой помощью с предварительным диагнозом инсультОпределение болезни. Причины заболеванияСимптомы болезни ФабриПатогенез болезни ФабриКлассификация и стадии развития болезни ФабриОсложнения болезни ФабриДиагностика болезни ФабриЛечение болезни ФабриПрогноз. ПрофилактикаИсточникиКлинические случаи
Болезнь Фабри известна с 1898 года, когда была впервые описана врачами Фабри и Андерсоном, имя которых и носит. Это очень редкое заболевание, распространенность его составляет от 1 : 117 000 до 1 : 476 000 населения.
Заболевание относится к лизосомным болезням накопления. При болезни Фабри возникает дефицит или снижение активности фермента лизосом α-галактозидазы А. Из-за этого не полностью расщепляются гликосфинголипиды. Промежуточные продукты обмена жиров (глоботриаозилцерамид и галабиозилцерамид) откладываются в различных органах и тканях, вызывая нарушение их функции. Прежде всего, накопление происходит в эндотелиальных и гладкомышечных клетках сосудов, клетках почек, сердечной мышцы, центральной нервной системе, клетках роговицы.
Причины
В основе заболевания лежит генетический дефект половой Х-хромосомы. В одном из участков этой хромосомы закодирована информация о ферменте α-галактозидазе А. Если возникает мутация, то это приводит к снижению количества этого фермента или уменьшению его активности.
Дефект имеет рецессивный характер наследования. Что это означает? Поскольку у мужского пола только одна Х-хромосома (а вторая – У), то у мальчиков при наличии патологической Х-хромосомы всегда развивается классическая картина заболевания. Больной мужчина обязательно передаст мутантную Х-хромосому всем своим дочерям без исключения (то есть в 100% случаев), а вот сыновья у него будут здоровыми.
У женщин две Х-хромосомы. Если одна из них мутантная, то у таких женщин возникают клинические проявления, но они менее выражены, позже развиваются и медленнее прогрессируют, почти всегда это атипичные формы болезни. Если же у женщины совпадут обе мутантные Х-хромосомы, одна от отца, другая – от матери (вероятность чего практически равна нулю, учитывая распространенность заболевания), тогда также развивается классическая картина болезни Фабри. Женщина может передать мутантную Х-хромосому как сыновьям, так и дочерям (при наличии одной мутантной Х-хромосомы вероятность составляет 50%).
Симптомы
Известно две формы заболевания:
- классическая: с началом в детском и подростковом возрасте, с полиорганным поражением;
- атипичная: с поздним началом и изолированным поражением одного органа (например, почек или сердца).
Хотя болезнь и передается по наследству, но ее проявления даже у членов одной семьи бывают различными. То есть совсем не обязательно, чтобы у одного больного одновременно наблюдались все симптомы заболевания. В большинстве случаев, при классической форме болезни Фабри симптомы со стороны различных органов и систем появляются постепенно, по мере прогрессирования заболевания.
При классической форме заболевание впервые дает о себе знать в детском возрасте (обычно до 10 лет).
Какие же клинические признаки поражения органов и систем при болезни Фабри? Это могут быть:
- кожные проявления: так называемые ангиокератомы. Это пятна на коже диаметром в несколько миллиметров разного цвета, от красного до синеватого. Пятна могут быть плоскими или несколько выступать над уровнем кожи. Они обычно располагаются на ягодицах, бедрах, в паху, в области пупка, на пальцах рук, на коленях и локтях, реже — на лице в виде «бабочки». При надавливании пятна не исчезают. Само по себе их наличие не доставляет беспокойства больному, кроме косметического дефекта. Появляются в детском возрасте и постепенно увеличиваются в размерах по мере взросления. Возможно появление подобных пятен на слизистой оболочке рта и конъюнктиве. По своему строению ангиокератомы представляют собой расширенные кровеносные сосуды, покрытые несколькими слоями кожи;
- полиневропатии: весьма характерный признак болезни Фабри. Проявляют себя следующим образом: больных беспокоит жгучая и сильная боль в конечностях. Боль возникает при небольшом болевом раздражении, при незначительном изменении температуры окружающей среды (особенно в ответ на теплую и горячую воду). Вместе с болями в кистях и стопах появляются ощущения жжения, покалывания, ползания мурашек (парестезии), которые подолгу не проходят, мучая больных. Помимо постоянных болей невропатического характера могут возникать болевые кризы: это мучительные боли в конечностях, отдающие в другие части тела, длящиеся от нескольких минут до нескольких дней, сопровождающиеся повышением температуры тела и СОЭ, не снимающиеся даже наркотическими препаратами. Кризы возникают в ответ на перемену погоды, физическую нагрузку, повышение температуры тела, стресс, прием алкоголя. Еще одним признаком полиневропатии при болезни Фабри является снижение или отсутствие потоотделения (гипо- или ангидроз);
- поражения центральной нервной системы: основной причиной «страдания» центральной нервной системы при болезни Фабри является отложение церамидов в стенках сосудов небольшого калибра. В результате этого возникают ишемические и геморрагические инсульты. Иногда болезнь дебютирует с развития инсульта. Все случаи инсультов у людей молодого возраста подозрительны в отношении болезни Фабри. Инсульты становятся причиной развития параличей, нарушения разборчивости и понимания речи, нарушения координации, судорожного синдрома. Если же недостаток кровоснабжения ткани мозга развивается постепенно, то тогда у больного исподволь ухудшается память, замедляются мыслительные процессы, возможно нарушение поведения и возникновение психических расстройств;
- поражение почек: начинается все с незначительной потери белка с мочой (в норме белок с мочой не выделяется). Это явление называется протеинурия. Постепенно протеинурия нарастает, с мочой теряется значительное количество белка, что приводит к отекам. Содержание белковых фракций в крови снижается. Постепенно почечные канальцы «забиваются», и развивается хроническая почечная недостаточность. И тогда организм не в состоянии избавиться от «шлаков», интоксикация нарастает. Кроме того, поражение почек приводит к возникновению артериальной гипертензии, то есть повышенного артериального давления, что может усугублять проявления заболевания со стороны центральной нервной системы и сердца. Единственный способ лечения терминальной почечной недостаточности – пересадка почки, либо таким больным показан постоянный гемодиализ. Довольно часто почечная недостаточность становится причиной смерти пациентов;
- поражение сердца: также может становиться причиной сокращения продолжительности жизни людей с болезнью Фабри. У большинства больных развивается утолщение (гипертрофия) стенок левого желудочка уже в молодом возрасте. Это состояние называется гипертрофической кардиомиопатией. Кровеносные сосуды сердца не в состоянии обеспечить необходимым количеством глюкозы такую толстую стенку миокарда. У таких больных появляются симптомы стенокардии, возможен даже инфаркт миокарда. Наряду с гипертрофией развивается фиброз, насосная функция сердца страдает, и появляется сердечная недостаточность. Нарушение питания стенок сердца приводит к возникновению нарушений сердечного ритма. Это может быть синдром слабости синусового узла, атриовентрикулярная блокада, мерцание предсердий. Нарушения сердечного ритма могут стать причиной внезапной смерти таких больных. Иногда при болезни Фабри развивается поражение клапанов сердца и крупных сосудов – чаще это митральная недостаточность и стеноз аорты;
- офтальмологические нарушения: выявляются у 70-90% больных. Возникает помутнение роговицы в виде завитков. Возможно постепенное формирование катаракты, что, наряду с поражением сосудов сетчатки, становится причиной тяжелой потери зрения;
- поражение желудочно-кишечного тракта: не очень частый симптом при болезни Фабри. Возможны тошноты, рвоты (следует иметь в виду, что они могут быть связаны с интоксикацией из-за хронической почечной недостаточности), послабление стула вплоть до диареи;
- костно-суставные проявления: периодические боли в суставах, повышение температуры и СОЭ симулируют суставные болезни. Со временем возможно развитие деформации межфаланговых суставов кистей и стоп, асептического некроза головки бедренной кости. В результате обменных нарушений постепенно кальций «вымывается» из позвонков, и развивается остеопороз;
- нарушения в системе свертывания крови: заключаются в развитии тромбозов периферических вен. Возможны спонтанные тромбоэмболии (например, тромбоэмболия легочной артерии), от которых больной может погибнуть;
- нарушение слуха и координации: при болезни Фабри больные часто жалуются на шум в ушах, у них постепенно снижается слух. Из вестибулярных нарушений характерны частые головокружения и, вследствие этого, неустойчивость при ходьбе.
Заболевание протекает таким образом, что к 30-40 годам у больных имеется целый «букет» из различных симптомов. Обычно к этому возрасту при классической форме болезни больной страдает от выраженной почечной недостаточности и имеет ряд сосудистых проблем со стороны центральной нервной системы или сердца.
Следует помнить, что существуют атипичные варианты течения заболевания, при которых наблюдается поражение одного органа или системы. При этом болезнь проявляет себя уже в зрелом возрасте, например, внезапным инсультом в 40 лет или сердечной недостаточностью неясного генеза.
Диагностика
Кроме разнообразных клинических проявлений для подтверждения диагноза болезни Фабри, необходимо определение активности лизосомального фермента α-галактозидазы А в культуре клеток кожных фибробластов, лейкоцитах, сыворотке крови, плазме, любом биоптате (кожи, почек и др).
Молекулярно-генетические методы диагностики позволяют обнаружить мутацию в определенном участке Х-хромосомы. Такие методы позволяют проводить пренатальную (дородовую) диагностику для исключения заболевания у плода в семьях, где наблюдалась мутация.
Лечение
С 2001 года в лечении заболевания успешно используется заместительная терапия рекомбинантными препаратами α-галактозидазы А. Это такие лекарственные средства, как Реплагал и Фабразим. Реплагал применяется внутривенно в дозе 0,2 мг/кг два раза в месяц, а Фабразим – по 1,0 мг/кг два раза в месяц. Оба препараты сопоставимы между собой по эффективности. На фоне применения этих препаратов удается добиться снижения выраженности болевого синдрома, регресса гипертрофии миокарда левого желудочка, стабилизировать функцию почек и предотвратить развитие хронической почечной и сердечной недостаточности.
Перспективы в лечении стоят за генной инженерией. Потенциально успешным, возможно, станет внедрение нормального гена (искусственно созданного), кодирующего α-галактозидазу А, в клетки организма человека, в частности, в костный мозг.
Наряду с заместительной терапией проводится симптоматическое лечение. Для уменьшения мучительных болей и парестезий используют антиконвульсанты (Карбамазепин, Габапентин, Прегабалин, Дифенин). Рекомендуется воздерживаться от физических нагрузок, избегать стрессов и изменений температуры. Также с обезболивающей целью используют пластыри и мази с лидокаином на зону локализации болевых ощущений.
Проблемы с почками и повышенное артериальное давление почечного генеза нивелируют путем применения ингибиторов АПФ (Рамиприл, Лизиноприл, Престариум, Энап) и блокаторов рецепторов ангиотензина II (Ирбесартан, Валсартан, Лосартан). В случаях, когда почечная недостаточность достигает терминальной стадии, показан гемодиализ или пересадка почки.
Учитывая склонность к тромбообразованию, таким больным назначают постоянно принимать Аспирин, Кардиомагнил или Клопидогрель, чтобы предотвратить осложнения в виде тромбозов и тромбоэмболий. Эти меры одновременно служат профилактикой развития инсультов.
При нарушениях сердечного ритма используют антиаритмические препараты.
Косметические дефекты (ангиокератомы) могут быть удалены с помощью лазерной терапии.
Таким образом, болезнь Фабри – это редкое наследственное заболевание с поражением многих органов. Чаще всего оно проявляет себя уже в детском возрасте, но возможны и более поздние формы (особенно у женщин). Если болезнь не лечить, она становится причиной выраженной почечной или сердечной недостаточности, инсульта, тромбоэмболии, от которых больной может погибнуть. Применение заместительной терапии позволяет предотвратить грозные осложнений и увеличить продолжительность и качество жизни таких больных.
Данное заболевание имеет второе название — болезнь Андерсена-Фабри. Называется из-за того, что ее открыли двое ученых. Данное заболевание имеет генетический характер и считается редким.
Описание
Суть болезни Фабри состоит в том, что в организме отсутствует такой фермент, как альфа-галактозидаза А. Этот элемент является важной составляющей практически всех клеток организма человека. Его функция состоит в расщеплении липидов. Болезни Фабри заключается в том, что человек может унаследовать ген, с которым передастся невозможность организма вырабатывать альфа-галактозидаза А. Такая особенность организма приведёт к тому, что в клетках начнёт накапливаться жирное вещество. Оно называется глоботриаозилцерамид. Накапливается он в лизосомах. Лизосомы — это отдел клеток, который собирает липиды. Жирное вещество накапливается, что приводит к деформации клеток. Далее повреждаются ткани и стенки сосудов. После чего появляются симптомы, характерные для болезни Фабри.
Если болезнь Фабри начинает прогрессировать и не оказывается своевременная медицинская помощь, то присутствует вероятность нарушения функций почек, сердца и головного мозга, появляется угроза жизни человека.
Группа риска
Кто находится в зоне риска такого недуга?
- Этим заболеванием могут заболеть как мужчины, так и женщины любой национальности.
- Проявления этого недуга может обозначиться в детском возрасте. Но, как правило, его диагностируют у подростков либо в период молодости. Существует статистика, что данный ген встречается редко.
Пути передачи
Как передается болезнь Фабри (МКБ-10 присвоила ей код E75.2)?
- Если в роду человека были родственники, которые имели неправильный ген, то существует вероятность передачи данного заболевания по наследству.
- Ген, который вызывает недуг Фабри, расположен на Х хромосоме (она является одной из двух составляющих, определения пола человека).
- Отличие мужчины и женщины состоит в том, что у первых присутствуют две хромосомы, а именно Х и Y. А женщины являются носителями двух одинаковых хромосом Х. Поэтому болезнь Фабри имеет свои отличия у мужчин и женщин. Также данная особенность влияет на способ передачи неправильного гена детям.
Особенности заболевания
Какие есть особенности болезни Фабри у мужчин?
- В связи с тем, что в организме парней присутствует одна Х-хромосома, то вероятность развития болезни выше, чем у женщин.
- Существует закономерность, что мужчина, организм которого поражён данным заболеванием, не может передать его своему сыну. Однако неправильный ген передаётся дочерям больного.
Особенности заболевания Фабри у женщин:
- У женщин в организме присутствуют две Х-хромосомы. Поэтому заболевание может проходить в более лёгкой форме и его признаки проявляются позже.
- Женщина может передать неправильный ген как дочерям, так и сыновьям. Вероятность того, что ребёнку достанется данное заболевание по наследству, составляет 50 %.
Как проявляется?
Особенностью данного заболевания является то, что оно имеет разную симптоматику у разных людей. У некоторых на протяжении всей жизни признаки носят слабый характер. А у кого-то проявляются в тяжёлых формах. Как говорилось выше, у представителей сильного пола признаки недуга можно заметить раньше, чем у девочек.
Статистика говорит о том, что диагноз «болезнь Фабри» ставится людям в возрастном промежутке от 30 до 45 лет. Хотя признаки заболевания были у человека раньше.
На это следует обратить внимание, если существует вероятность передачи данного заболевания по наследству.
Болезнь Фабри. Симптомы
Какие есть признаки заболевания?
- Болевые ощущения в конечностях. Как правило, они возникают при физических нагрузках и в жаркую погоду.
- Бордовая сыпь на коже. Как правило, она появляется в зоне пупка и до колен.
- Нарушается функция потоотделениям, что влияет на плохое самочувствие человека в жаркую погоду.
- Видоизменяется роговая оболочка глаза, при этом зрение остаётся нормальным.
В некоторых случаях врачи путают болезнь Фабри (фото с проявлениями не раз видели врачи) с переутомлением или усталостью человека. Через определённое время, если заболевание не лечится и продолжает развиваться в организме, то к вышеперечисленным признакам добавляются еще и другие.
Симптомы развития заболевания
Какие есть признаки у человека, у которого диагностирована болезнь Фабри? Признаки развития данного недуга следующие:
- Утомление организма, снижение трудоспособности.
- У человека присутствуют боли в животе, ускоряется процесс переваривания пищи, его начинает тошнить, появляется диарея.
- Возникают болевые ощущения в голове.
- Ухудшение слуха.
- Появляются отеки ног.
- Болевые ощущения в грудной клетке, учащается сердцебиение.
Осложнения
При дальнейшем прогрессировании болезни Фабри могут возникнуть следующие осложнения:
- Дестабилизируется работа почек. Из организма начинает выводиться белок. Развивается почечная недостаточность.
- Поражение сердечной системы. Может измениться его форма, возникнуть аритмия.
- Недостаточное питание головного мозга, что повышает вероятность возникновения инсульта.
Методы диагностики
Как определить, что у человека болезнь Фабри? Диагностика требуется. Причем чем раньше ее провести, тем лучше. Для диагностики у больного берут анализы крови, мочи, плазмы. Данный материал исследуется для выявления необходимых элементов. Только после проведенной диагностики назначается лечение.
Болезнь Фабри является серьёзным нарушениям нормального функционирования человеческого организма. Постоянные боли, и дискомфорт могут привести к возникновению депрессии, плохому самочувствию, нежеланию работать и учиться.
Дефектный ген может спровоцировать то, что у человека возникнет инсульт, инфаркт и прочие осложнения, которые представляют опасность жизни. Несколько лет назад существовала статистика, что люди, которые болеют Фабри, в среднем живут на двадцать лет меньше других.
В настоящее время достижения современной медицины позволяют диагностировать данное заболевание, а также лечить его. Поэтому не медлите!
Болезнь Фабри. Лечение и профилактика
Пациенту сначала назначаются болеутоляющие средства. Они облегчают состояние больного. Другим направлением лечения является восполнение в организме фермента, который не вырабатывается из-за испорченного гена. Называется оно ферментно-заместительная терапия.
Существует мнение, что если диагностировать заболевание на ранней стадии и применить пациенту данное лечение, то можно одолеть болезнь Фабри. Профилактика данного недуга отсутствует, так как оно передается по наследству. Единственным способом избежать рождения ребенка с этим заболеванием является диагностирование культивируемых амниотических клеток активности a-галактозидазы А.
Самым лучшим вариантом для носителей этой болезни является его раннее диагностирование. Для этого не нужно пускать на самотек жалобы ребенка о присутствии болевых ощущений в кистях рук или ног. Особенно если известны случаи, что в роду кто-то из родственников страдал от данного недуга. Современная медицина позволяет оказать помощь людям, у которых присутствует болезнь Фабри в организме. Поэтому лучше обратиться к специалисту, сделать обследование организма и начать своевременное лечение.
Как помочь ребенку?
При вероятности присутствия данного заболевания у ребенка и подозрении на то, что у него проявляются определенные симптомы болезни, следует обратиться к педиатру. На приеме у врача необходимо поделиться своими опасениями о возможности передачи данного заболевания по наследству. Также нужно рассказать, что данный недуг присутствовал у родственников ребенка. Далее врач осмотрит больного, назначит необходимые анализы для выявления дефицита альфа-галактозидаза А. Также педиатр выдаст направление к генетику. Тот проанализирует полученные данные и назначит лечение, которые стабилизирует состояние пациента.
Что делать взрослому? План действий
При обнаружении симптомов Фабри во взрослом возрасте следует записаться на прием к терапевту и рассказать ему о симптомах болезни. Не стоит запускать заболевание и заглушать болевые ощущения средствами. Необходимо провести лечение, связанное с восполнением отсутствующего фермента и тем самым наладить работу организма.
Если заболевание протекает в классической форме, то его первые признаки дадут о себе знать до достижения ребенком десятилетнего возраста. Далее болезнь начинает прогрессировать. К достижению человеком сорока лет он имеет целый комплекс заболеваний, связанных с различными органами и системами. Сюда относятся и кожные высыпания, и проблемы с сердцем, почками и желудком.
Небольшое заключение
Теперь вы знаете, что представляет собой болезнь Фарби. Мы описали все признаки заболевания. Также была затронута тема диагностики и лечения данного недуга. Помните, что это очень серьезное заболевание. Поэтому нужно сразу же обратиться к квалифицированному доктору, чтобы тот определил болезнь и назначил необходимое лечение.
Похожие статьи
Болезнь Фабри (Андерсона-Фарби) относится к тому виду генетических заболеваний, с которыми врачи в своей практике сталкиваются крайне редко.
Из-за мутации гена определённый вид жиров гликосфинголипидов накапливается в клетках организма и вызывает нарушение работы почек, органов зрения, сердца, нервной системы, кожных покровов и т.д. При своевременной диагностике и правильно проведенной заместительной терапии можно приостановить ее развитие и увеличить продолжительность жизни пациента.
Болезнь впервые описана в 1898 г врачами Джон Фабри (Германия) и Вильямом Андерсоном (Великобритания) независимо друг от друга. Она имеет низкую частотность, но отмечается во всех этнических группах. По причине низкой частотности ранняя диагностика затруднена. Самая распространённая её форма – атипичная с поражением какого-либо одного органа.
В России на сегодня официально зарегистрировано 20 пациентов с этим диагнозом. Классификация по МКБ-10 E75.2 (ILDS E75.25).
Болезнь Фабри – дефицит фермента
Причиной болезни Фабри является снижение или отсутствие фермента альфа-галактозидазы, вызванное мутацией гена, который отвечает за его активность. При недостатке фермента в лизосомах клетки накапливаются промежуточные продукты обмена, нарушается метаболизм мембранных гликосфинголипидов.
При скоплении продуктов обмена в малых и крупных сосудах в организме начинают постепенно развиваться патологические процессы. Первыми признаками являются характерные кожные высыпания пурпурного цвета, приступы лихорадки и острая боль в руках и ногах – в первую очередь страдают кисти и стопы.
Единственным видом лечения на сегодня является фермент-заместительная терапия.
Причины
Поскольку синдром Фабри наследственное заболевание, важно понять, каким образом это происходит. Это Х-сцепленное заболевание с доминантным признаком наследования, хотя ранее оно считалось рецессивным. Его вызывает дефект Х-хромосомы, что чаще отмечается у мальчиков.
Обусловлено это тем, что у мужчин X и Y хромосомы, а у женщин две X-хромосомы. Женщина передает мутировавшую хромосому и дочери, и сыну; мужчина – только дочерям.
Все дочери больного мужчины и здоровой женщины будут носительницами бракованного гена, а сын может быть полностью здоров и не передавать его потомкам. Мальчик может получить его только от матери, при чем болезнь будет развиваться по классическому сценарию.
При наличии мутировавшей хромосомы у женщины клинические проявления болезни менее выражены, она развивается позже, и прогрессируют значительно медленнее, чем у мужчин. В большинстве случаев развивается по атипичному сценарию.
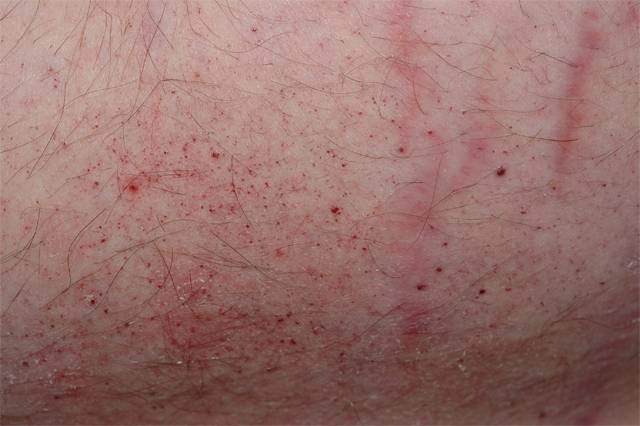
Симптомы
Болезнь начинает развиваться ещё во внутриутробный период, так как обусловлена наследственным фактором. Но поскольку она прогрессирует медленно, первые признаки отмечаются у мальчиков в возрасте 6-12 лет, иногда еще позже. У женщин признаки болезни появляются значительно позже, иногда после 50 лет.
Первичные симптомы у детей проявляются в виде специфической сыпи пурпурного цвета (ангиокератомы) в области пупка или нижней части живота, нарушением потоотделения (гипогидроз, ангидроз) и болей в кистях и стопах, которые отмечаются при жаркой погоде, повышенной температуре тела.
У взрослых отмечается поражение только какого-то одного органа. При этом диагностика болезни затрудняется сходностью симптомов с распространёнными заболеваниями. Признаки ишемического инсульта, сердечной, почечной недостаточностей или невропатические боли могут оказаться проявлением симптома Фабри.
Клиническими проявлениями патологий органов и систем могут стать:
- Кожные высыпания ангиокератомы. Пятна размером в несколько миллиметров пурпурно-красного цвета. Появляются в области пупка, паховой области, на пальцах, коленях, на лице и т.д. По структуре это увеличенные участки кровеносных сосудов с несколькими образовавшимися слоями кожи. Не доставляют особого беспокойства пациенту, кроме эстетического. При надавливании на них не исчезают и не болят. Увеличиваются с ростом ребенка.
- Полиневропатии. Один из самых характерных признаков заболевания. Проявляется в виде жгучей боли в конечностях, которая появляется при температурных перепадах и незначительных воздействиях. Боли сопровождаются длительным жжением, покалыванием, «мурашками», может отдавать и в другие части тела. Длятся до нескольких дней. Боль сильная и не снимается сильными анальгетиками.
- Нарушение потоотделения. Снижается или полностью отсутствует потоотделение при температурных перепадах (реакция на жару) и при повышении температуры тела.
- Заболевания ЦНС. Проявляются в виде ишемических или геморрагических инсультов, вызванных отложениями церамидов на стенках сосудов. Как правило, этот вид проявления болезни Фабри характерен для людей молодого возраста и является показанием для более глубокого обследования.
- Нарушение кровообращения. Проявляется в развитии тромбов в периферических венах, тромбоэмболии, которая может стать причиной смерти пациента.
- Костно-суставные изменения. Боли в суставах, периодическое повышение температуры, высокий показатель СОЭ приводят к деформации суставов кистей и ступней, заболеваниям головки бедренной кости (некроз). Из-за недостатка кальция в костях позвоночника развивается остеопороз.
- Патологии почек. Первый признак – выделение белка с мочой (протеинурия). Постепенно количество белка в организме уменьшается, что приводит к появлению отёков. В организме возрастает содержание токсинов и развивается хроническая почечная недостаточность. Поражение почек приводит к повышению артериального давления, что усиливает нагрузку на сердце и ЦНС. Лечением почечной недостаточности является либо пересадка почки, либо постоянное проведение гемодиализа. Во многих случаях это заболевание становится причиной смерти пациента.
- Заболевания сердца. У большинства больных болезнь Фабри вызывает утолщение стенок левого желудочка (гипертрофию) еще в молодом возрасте. Гипертрофическая кардиомиопатия заключается в том, что кровеносные сосуды сердца не в состоянии доставить необходимое количество глюкозы в ткани миокарда. Это приводит к стенокардии или к инфаркту миокарда. С нарушениями сердечного ритма связаны митральная недостаточность или стеноз аорты.
- Слух и координация движений. У больных синдромом Фабри часто отмечается шум в ушах, вестибулярные нарушения. Наступает ранняя глухота, отмечаются нарушения в походке.
- Офтальмологические проблемы. Проявляются в помутнении роговицы в виде завитков. Способствует формированию катаракты, дистрофии сетчатки и приводит к слепоте. Проблема отмечается в 70-90% случаев больных синдромом Фабри.
Формы развития заболевания
Различают три формы болезни Фабри:
- Классическая. Первые характерные признаки заболевания ярко выражены в раннем возрасте – до 20 лет.
- Атипичная. Проявляется в более позднем возрасте в виде поражения сердца, почек, головного мозга. Диагностика значительно осложнена.
- Женская. Проявляется значительно позже, чем у мужчин. Симптомы слабо выражены, форма течения самая лёгкая.
Диагностика
Сложность в постановке диагноза заключается в том, что врач изначально должен заподозрить болезнь Фабри и назначить проведение необходимых анализов. Поскольку случаи заболевания крайне редкие, в основном лечат пациента по имеющимся симптомам без уточнения окончательного диагноза.
Для диагностики проводятся лабораторные и инструментальные исследования. Они включают:
- Анализ на активность альфа-галактозидазы. Для исследования делают забор крови, берут засохшие пятна крови, проводят биопсию почек. У мужчин активность фермента явно снижена; у женщин показатели могут находиться в норме или отмечаться незначительное снижение.
- Анализ сфинголипидов. Проводится количественный анализ в плазме и сухих пятнах крови. Высокое содержание сфинголипидов подтверждает заболевание. Скрининг проводится также для контроля результатов проводимого лечения.
- Секвенирование ДНК. Исследуется ген GLA на предмет мутационных изменений в структуре. Это один из самых точных способов диагностики синдрома Фарби, в частности у женщин.
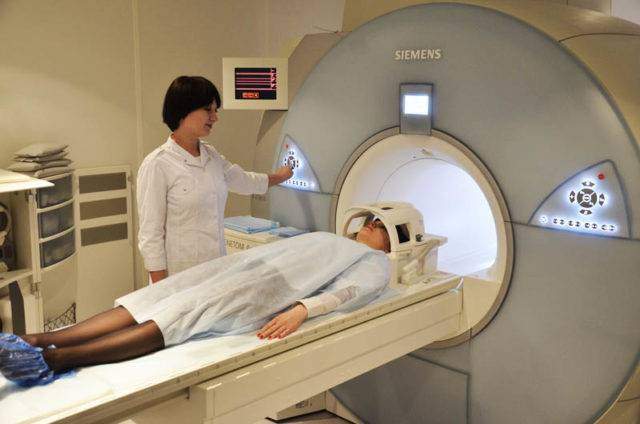
- МРТ головного мозга. При исследовании у больных отмечается изолированное поражение заднего бугорка таламуса, выявляются сосудистые мальформации (аномалии, образования).
- В качестве вспомогательных исследований проводятся ЭКГ, Эхо-КГ, МРТ сердца. В результате отмечаются изменения (гипертрофия) левого желудочка.
Методы лечения
Самым эффективным и единственным на сегодня способом лечения является фермент-заместительная терапия. Этот метод позволяет заменить пациенту фермент, которого не хватает в организме у больного. Препараты начали успешно использовать в начале 2000 годов. Их приём позволяет значительно улучшить самочувствие пациента, однако не приводит к полному исцелению.
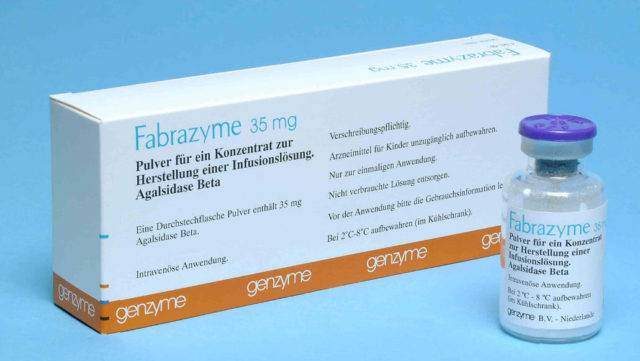
Фермент-заместительная терапия
Для этого используются рекомбинантные препараты альфа-галактозы. Сегодня успешно применяют Реплагал и Фабразим. Оба вводятся внутривенно, с определённой периодичностью. Процесс инвазии занимает около получаса. Препараты стоят очень дорого, поэтому лечение больных синдромом Фабри оплачивается из федерального бюджета.
Они помогают облегчить состояние больного и частично восстановить утраченные ранее функции почек, снизить болевой синдром, приостановить развитие изменений в левом желудочке сердца, предотвратить хроническую почечную и сердечную недостаточность. Улучшения в состоянии пациента наступают быстрее в случае, если лечение начало проводиться при первых симптомах заболевания.
Симптоматическое лечение
Для улучшения самочувствия больного используют препараты, способные устранить патологические проявления болезни. Для уменьшения болевого синдрома и парестезий назначают антиконвульсанты, кортикостероиды, анальгетики и нестероидные противовоспалительные препараты. Для снятия боли местно рекомендовано использование пластырей и мазей, в состав которых входит лидокаин.
При проблемах с почками и артериальной гипертензии назначается ингибиторы АПФ и блокаторы ангиотензина II. В термальной стадии периодично проводят гемодиализ или трансплантацию почки.
При повышенной свёртываемости крови для предотвращения образования тромбов и тромбоэмболии а также снижения риска инсульта назначают прием аспирина или кардиомагнила.
Ангиокератомы (пятна на коже) удаляют при помощи лазера.
Прогноз
При ранней постановке диагноза у пациента с синдромом Фабри прогноз благоприятный. Фермент-заместительная терапия показывает хорошие результаты. Первые процедуры инфузии проводятся стационарно для того, чтобы отследить переносимость препарата, при необходимости скорректировать дозировку. В дальнейшем показано амбулаторное лечение.
Поздно диагностированная болезнь у мужчин прогрессирует значительно быстрее, чем у женщин. Отказ от лечения приводит к смерти от почечных, сердечно-сосудистых и неврологических заболеваний.
Профилактика
Как и другие генетические заболевания, болезнь Фабри не имеет методов профилактики. Возможно только предупреждение появления ребёнка с этой наследственной мутацией. Для этого рекомендована медико-генетическая консультация и проведение пренатальной диагностики. Специалист-генетик в ходе консультации оценивает анамнез уже на этапе планирования беременности. При необходимости назначается ДНК-диагностика для выявления мутаций в гене GLA у будущих родителей. Врач даёт рекомендации исходя из риска вероятности мутации у будущего ребёнка.
Диагностика у плода проводится на основе анализа амниотических клеток или биопсии хориона. При положительном тесте родители решают, продолжать или прерывать беременность. Появление на свет больного ребёнка зависит только от выбора родителей.
Используемые источники:
- https://probolezny.ru/bolezn-fabri/
- https://doctor-neurologist.ru/bolezn-fabri-prichiny-simptomy-lechenie
- https://www.syl.ru/article/292490/bolezn-fabri-prichinyi-simptomyi-lechenie
- https://onevrologii.ru/nasledstvennye-zabolevaniya/bolezn-fabri






